Original
Muerte súbita cardíaca asociada a enfermedades mitocondriales
Sudden cardiac death in mitochondrial diseases

DOI:
10.59457/cmf.2022.25.01.org04
CITAR EL ARTÍCULO
Tenhaeff Lackschewitz P, Molina Aguilar P, Mancheño Franch N, Zorio Grima E. Muerte súbita cardíaca asociada a enfermedades mitocondriales. Cuad Med Forense. 2022; 25(1):45-62. DOI: 10.59457/cmf.2022.25.01.org04Cuad Med Forense. 2022; 25(1):45-62
Enviado: 16.06.22 | Revisado: 30.06.22 | Aceptado: 17.07.22
Resumen
Las enfermedades mitocondriales (EM) constituyen un grupo heterogéneo de trastornos genéticos que resultan de la disfunción de la vía común final del metabolismo energético. La mutación responsable se puede encontrar en genes del ADN nuclear o del ADN mitocondrial lo que implica diferencias en la forma de presentación y en el patrón de herencia. Estas enfermedades suelen tener una presentación multisistémica, con afectación especial de tejidos con un alto requerimiento energético oxidativo (músculo cardíaco, esquelético y cerebro, entre otros). Las causas de muerte más frecuentes en las EM están relacionadas con la severidad de la afectación clínica, si bien es también posible que se produzca una muerte súbita cardíaca (MSC) en pacientes con clínica leve y en asintomáticos. El objetivo de este trabajo de revisión es la identificación de los casos publicados de MSC en EM (conocida en vida o diagnosticada en estudios post mortem), así como un análisis sistemático de los procesos patológicos macroscópicos y microscópicos subyacentes. Existen pocos casos documentados (27) aunque la falta de rasgos fenotípicos cardíacos marcados en algunos casos, la baja especificidad de los mismos, la escasa información clínica personal y familiar en el momento de la autopsia y el bajo nivel de alerta para el diagnóstico de estas enfermedades plantean la posibilidad de que exista un infradiagnóstico post mortem, pasando muchos casos desapercibidos. Sin duda, un correcto diagnóstico requiere de un alto nivel de sospecha y un estudio multidisciplinar con estudios anatomopatológicos y cardiológicos especializados, tanto en el fallecido como en los familiares en riesgo para confirmar o descartar la enfermedad a fin de ajustar los seguimientos y tratamientos en caso de ser necesarios y dar un consejo genético certero.
Palabras clave: Muerte súbita cardíaca; Enfermedad mitocondrial; ADN mitocondrial; Miocardiopatía mitocondrial.
Abstract
Mitochondrial diseases (MD) constitute a heterogeneous group of genetic disorders that result from the dysfunction of the final common pathway of energy metabolism. The responsible mutation can be found in genes encoded in nuclear DNA or in mitochondrial DNA, causing differences in the clinical presentation and inheritance pattern. Most of these diseases have a multisystemic impact, mainly affecting tissues with a high oxidative energy requirement (heart muscle, skeletal muscle, and brain, for instance). Although death in DM is usually related to the clinical severity of the disease, sudden cardiac death (SCD) is possible also in patients with mild manifestations or even in asymptomatic individuals. This review aims to identify reported cases of SCD in MD (either diagnosed in life or at post mortem studies) and comprehensively analyze the macroscopic and microscopic pathological features described. There are only few documented cases (27), but the lack of notable cardiac phenotypic features in some cases, their low specificity, the scarce clinical personal and family information when the autopsy is performed and the little awareness of these diseases among doctors raise the suspicion that MD are infradiagnosed and many cases remain overlooked. Definitely, a correct diagnosis requires a high level of suspicion and a multidisciplinary approach with pathological and cardiological specialized tests both in the deceased proband and in the at-risk relatives to confirm or discard DM in order to tailor the follow-up, treatment and genetic counselling.
Key words: Sudden cardiac death; Mitochondrial disease; Mitochondrial DNA; Mitochondrial cardiomyopathy.
INTRODUCCIÓN
La muerte súbita (MS) es aquella que tiene una causa natural, no traumática y aparece de forma inesperada en un corto periodo de tiempo tras el inicio de los síntomas (1). La MS se clasifica como muerte súbita cardíaca (MSC) si se conoce una enfermedad cardíaca potencialmente fatal, congénita o adquirida, cuando en la autopsia se encuentra una anomalía cardíaca o vascular como la probable causa de muerte o si tras una completa se han descartado las causas extracardíacas en una autopsia completa con estudios complementarios (histología, toxicología, microbiología) y, por tanto, una arritmia maligna es la causa más probable de muerte (1). Entre las causas de MSC hay que diferenciar tres grandes grupos: las isquémicas, las miocardiopatías y el síndrome de muerte súbita arrítmica (SMSA). La cardiopatía isquémica es la causa más frecuente de MSC a partir de los 35 años, sin embargo, las miocardiopatías, canalopatías y el SMSA son porcentualmente más importantes en edades más tempranas (1).
Las enfermedades metabólicas hereditarias son un grupo muy amplio de enfermedades genéticas, muy heterogéneas, que se caracterizan por una alteración del metabolismo. En este amplio grupo de enfermedades se incluyen las enfermedades mitocondriales (EM) debidas a mutaciones en genes que codifican proteínas de función mitocondrial y resultan en una disfunción de la cadena de transporte de electrones y la fosforilación oxidativa (2). La prevalencia de las enfermedades mitocondriales se estima en 1 por cada 5.000 nacidos vivos (3). Las mutaciones de las enfermedades mitocondriales pueden ocurrir tanto en el ADN nuclear (ADNn) como en el mitocondrial (ADNmt). El mecanismo de herencia cuando las mutaciones se localizan en el ADNn es, en su gran mayoría, autosómico recesivo, a veces ligado al cromosoma X y, en raras ocasiones, también autosómico dominante. Sin embargo, cuando las mutaciones afectan a genes localizados en el ADNmt presentan un patrón de herencia matrilineal en el que sólo las mujeres pueden transmitir la enfermedad a su descendencia (3).
En la mitosis, el ADNn se duplica y se divide de forma que las células hijas tienen el mismo material genético que la célula madre, sin embargo, el ADNmt no tiene esta forma de división. Cada célula tiene muchas mitocondrias y dentro de cada mitocondria hay varias copias de ADNmt, de 100 a 10.000 copias dependiendo del tejido analizado. Cuando la célula se prepara para dividirse crece y aumenta su número de mitocondrias. En telofase, al dividirse en dos, estas mitocondrias se reparten de forma aleatoria entre ambas células hijas. En pacientes con EM podría ocurrir que, en algunas células, el ADNmt fuera enteramente sano o mutado, las células hijas serán sanas o mutadas en su totalidad, en este caso hablamos de homoplasmia. Sin embargo, si en una célula de ese paciente coexiste ADNmt mutado y sano (wild type), las células hijas tendrán diferentes porcentajes de ADNmt mutado. Debido a la aleatoriedad en la división de las mitocondrias, el porcentaje de ADNmt mutado puede variar entre diferentes órganos y tejidos, existiendo riesgo de que pacientes con baja heteroplasmia en sangre pueda tener niveles altos en otros tejidos. Por tanto, la expresión fenotípica será variable en cada sujeto e incluso en cada tejido, dependiendo del número final de copias de ADNmt mutado (carga absoluta de la mutación), del porcentaje de mitocondrias con ADNmt mutado (heteroplasmia, carga relativa de la mutación) y de algunos factores nucleares característicos de cada tejido.
Este concepto es muy importante porque explica, en parte, la gran heterogeneidad clínica observada en las enfermedades mitocondriales. Se piensa que el grado de heteroplasmia contribuye a determinar el fenotipo clínico. La célula tiene que superar cierto umbral (es decir, porcentaje de copias de ADNmt mutado) para que se produzca la alteración bioquímica. Se han propuesto diferentes porcentajes umbral, pero como regla general se suele necesitar más de un 50-60% para mostrar alteraciones relevantes (4,6,7). Sin embargo, se han informado estados clínicos de enfermedad cuando los niveles de ADNmt mutante eran tan bajos como un 10% (6).
En general, los pacientes con altos niveles de heteroplasmia presentan enfermedades muy discapacitantes que suelen estar diagnosticadas y llevar a una muerte temprana. Sin embargo, si los niveles de heteroplasmia son bajos pueden ser asintomáticos. De hecho, las mutaciones puntuales en el ADNmt son bastante frecuentes pero una gran mayoría presentan baja heteroplasmia y no tienen trascendencia clínica (8).
Dado que las mitocondrias de un cigoto proceden del óvulo y no del espermatozoide que le da origen, sólo las madres pueden transmitir la enfermedad a su descendencia, independientemente de que sean niños o niñas. Esta forma de herencia se denomina matrilineal y es típica de las enfermedades mitocondriales por mutaciones en ADNmt (3–6).
En los últimos años se ha destacado la importancia de detectar anomalías cardíacas estructurales y funcionales subclínicas en pacientes con EM conocidas, no reconocidas en el pasado. La historia natural de la cardiopatía subclínica no se conoce con claridad, pero se han descrito un mayor número de casos de MSC en los últimos años (9). Debido a esto, hoy en día es práctica clínica rutinaria monitorizar la función cardíaca con electrocardiogramas y ecocardiogramas regulares (10), ya que son pruebas accesibles, no invasivas y de bajo coste que podrían detectar anomalías en al menos un porcentaje de estos pacientes. Los principales síndromes con afectación cardíaca se detallan en la Tabla 1.
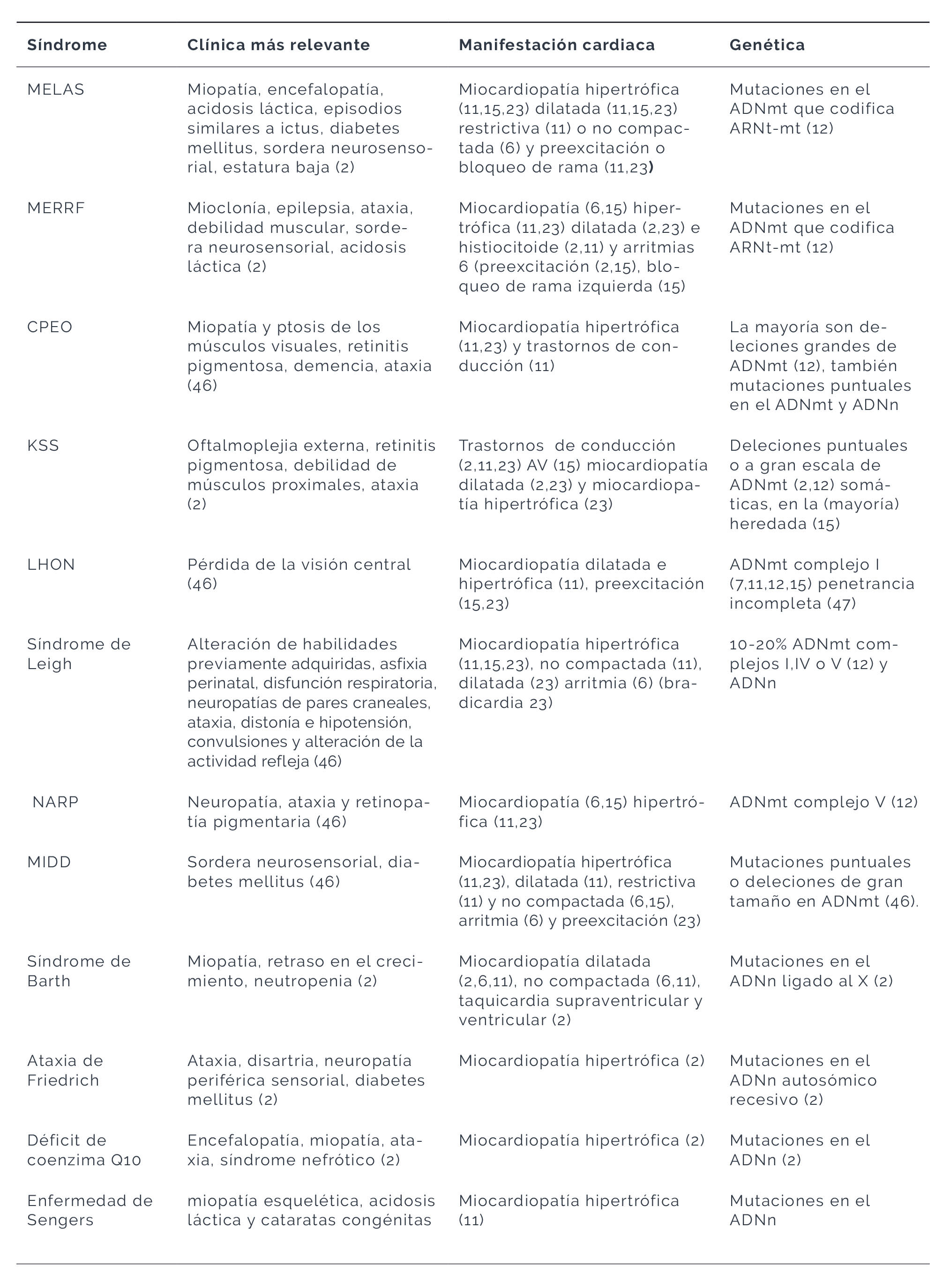
Tabla 1. Principales síndromes con afectación cardíaca
Este trabajo pretende hacer una revisión de los casos publicados de MSC en personas con EM (conocida o no en vida) y profundizar en los procesos patológicos de dicha enfermedad. Es de especial importancia ya que, a pesar de que no comprenda un elevado número de casos, son pacientes de difícil diagnóstico y con un gran impacto social y familiar. En casos de MSC sin diagnóstico previo de EM, su correcto diagnóstico post mortem no sólo esclarece la causa de muerte del individuo y puede ser de ayuda para mejorar la estratificación de riesgo en este escenario clínico, sino que también tiene mucha relevancia para orientar los estudios genéticos necesarios y proporcionar consejo genético, seguimiento y tratamiento a los familiares del paciente que resulten afectados.
MATERIAL Y MÉTODOS
La búsqueda de información para esta revisión se ha realizado a través de la base de datos de MEDLINE, mediante el motor de búsqueda de libre acceso PubMed. El algoritmo de búsqueda utilizado ha sido mediante las palabras clave: (sudden death) OR (sudden cardiac death) AND (mitochondrial cardiomyopathy) OR (mitochondria) OR (mitochondrial DNA) localizadas en el título o en el abstract. La selección se ha restringido a artículos publicados entre el 2001 y 2021 en inglés o castellano. De los 114 artículos obtenidos, se ha revisado el abstract y se han elegido aquellos que se adaptan al objetivo del presente trabajo. De entre los que han resultado apropiados para este trabajo se han recopilado artículos presentes en sus referencias que hayan sido de especial relevancia.
RESULTADOS
1. MIOCARDIOPATÍA MITOCONDRIAL
La miocardiopatía mitocondrial es un trastorno miocárdico, sin enfermedad coronaria concomitante, enfermedad valvular, cardiopatía congénita o hipertensión arterial, caracterizada por una estructura y/o función miocárdica anormal, secundarias a alteraciones en genes mitocondriales que resultan del deterioro de la función de la cadena respiratoria mitocondrial (6,11).
1.1. Fenotipos macroscópicos
Miocardiopatía hipertrófica (MCH). El remodelado hipertrófico es la forma más común de miocardiopatía en todas las enfermedades mitocondriales (alrededor del 40-50% de los pacientes) (11,12). Imita a la MCH, por lo que es considerada una fenocopia de ésta. Existen, no obstante, algunas diferencias fenotípicas reseñables. La probabilidad de progresión a dilatación ventricular e insuficiencia cardíaca es mayor que en la MCH sarcomérica, sin embargo, la obstrucción del tracto de salida del ventrículo izquierdo se observa más raramente en las EM (2,12,13).
Miocardiopatía dilatada (MCD). La mayor parte del remodelado hacia dilatación ventricular y disfunción sistólica en las EM se debe a la progresión desde un fenotipo previo hipertrófico (2,12). Se ha descrito MCD en KSS (Kearns-Sayre syndrome), MELAS (mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes) y MERRF (myoclonic epilepsy with ragged red fibres) (2).
Miocardiopatía no compactada o hipertrabeculación del ventrículo izquierdo. Este tipo de miocardiopatía es raro en las miocardiopatías mitocondriales. Sin embargo, entre los pacientes que presentan miocardiopatía no compactada, las enfermedades mitocondriales son altamente prevalentes (2).
Miocardiopatía histiocitoide. Muy raramente se ha descrito en esta miocardiopatía enfermedad mitocondrial tanto de herencia autosómica recesiva como ligada al cromosoma X y matrilineal (2,14).
Miocardiopatía restrictiva. Es una presentación infrecuente en la EM, pero se ha informado en asociación con sordera y diabetes heredadas de la madre debido a la mutación m.3243A>G en el gen MT-TL1 y como único hallazgo clínico en un sujeto con la mutación m.1555A>G en el gen 12S rARN (12).
Corazón estructuralmente normal. Las EM pueden ocasionar trastornos de conducción potencialmente letales en ausencia de cardiopatía estructural. De hecho, algunos de los casos de MSC por EM descritos en la literatura han cursado como SMSA y la confirmación diagnóstica ha sido posible tras el estudio genético (9,15,16,17).
1.2. Hallazgos microscópicos
Hay pocos casos de miocardiopatías mitocondriales con MSC documentados a nivel histológico y sus rasgos más característicos son (ver tabla 2, 2 bis y tabla 2 ter):
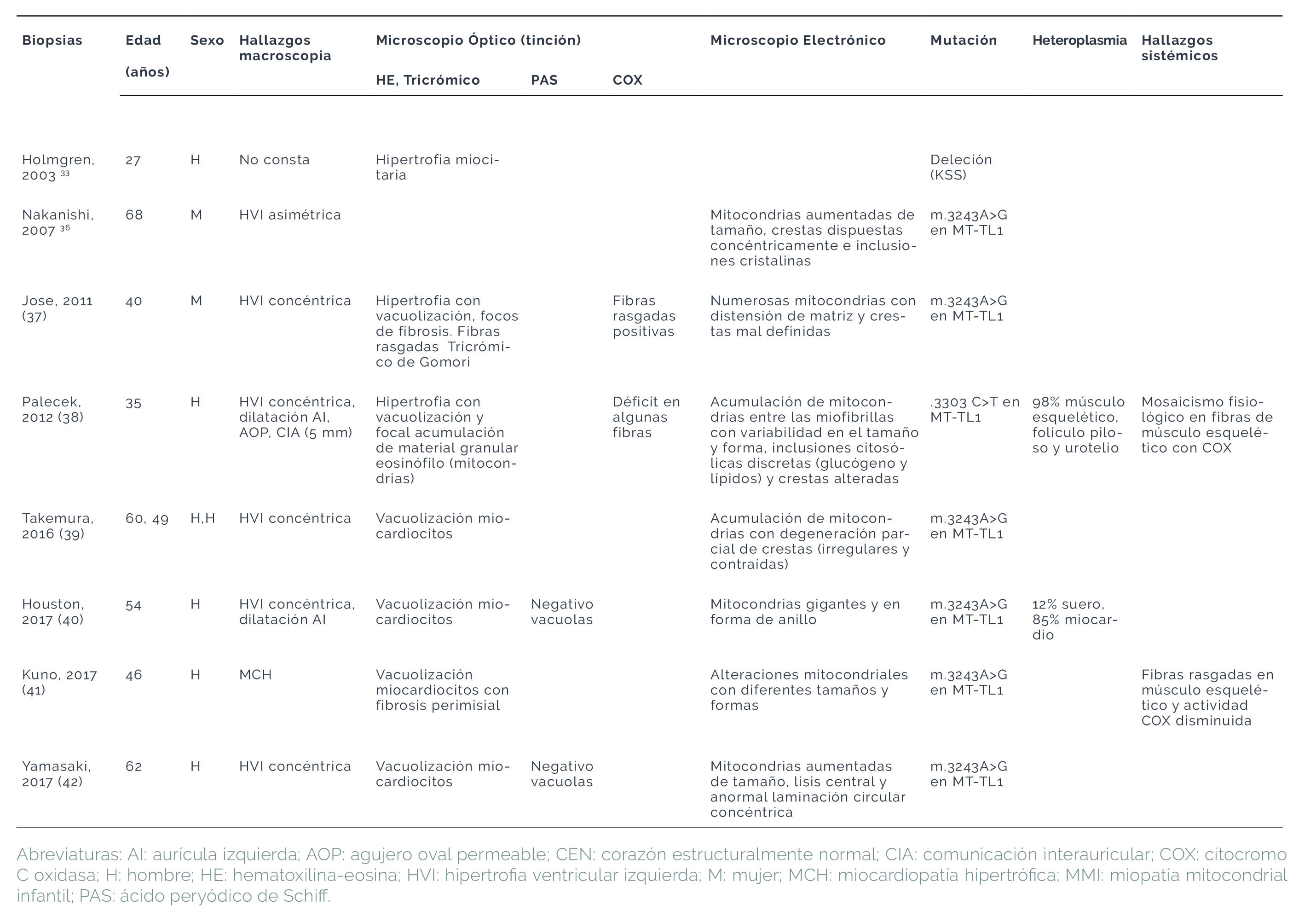
Tabla 2. Hallazgos histopatológicos descritos en los casos de cardiopatías mitocondriales. Casos derivados de biopsias cardíacas.
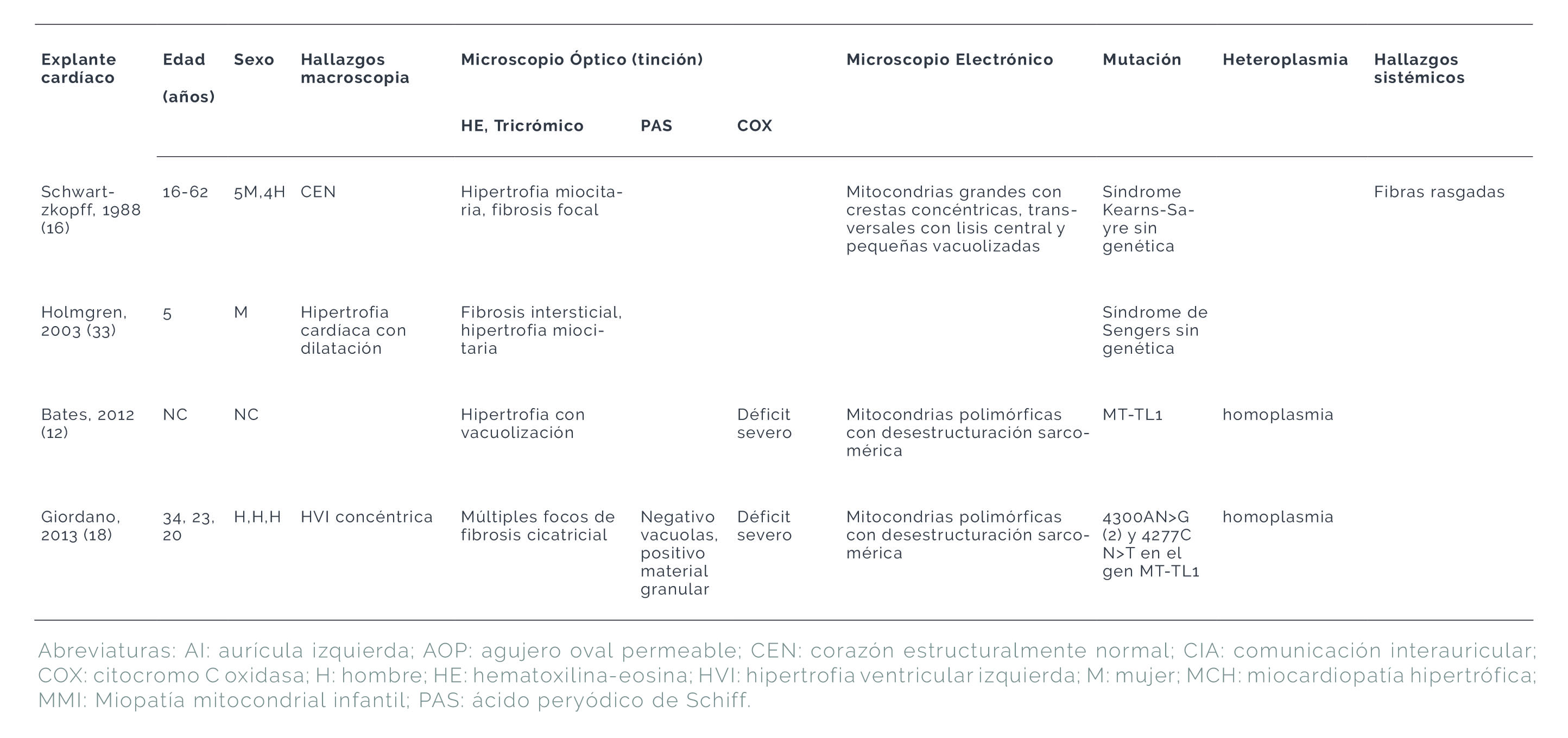
Tabla 2 bis. Hallazgos histopatológicos descritos en los casos de cardiopatías mitocondriales. Casos derivados de explantes cardíacos
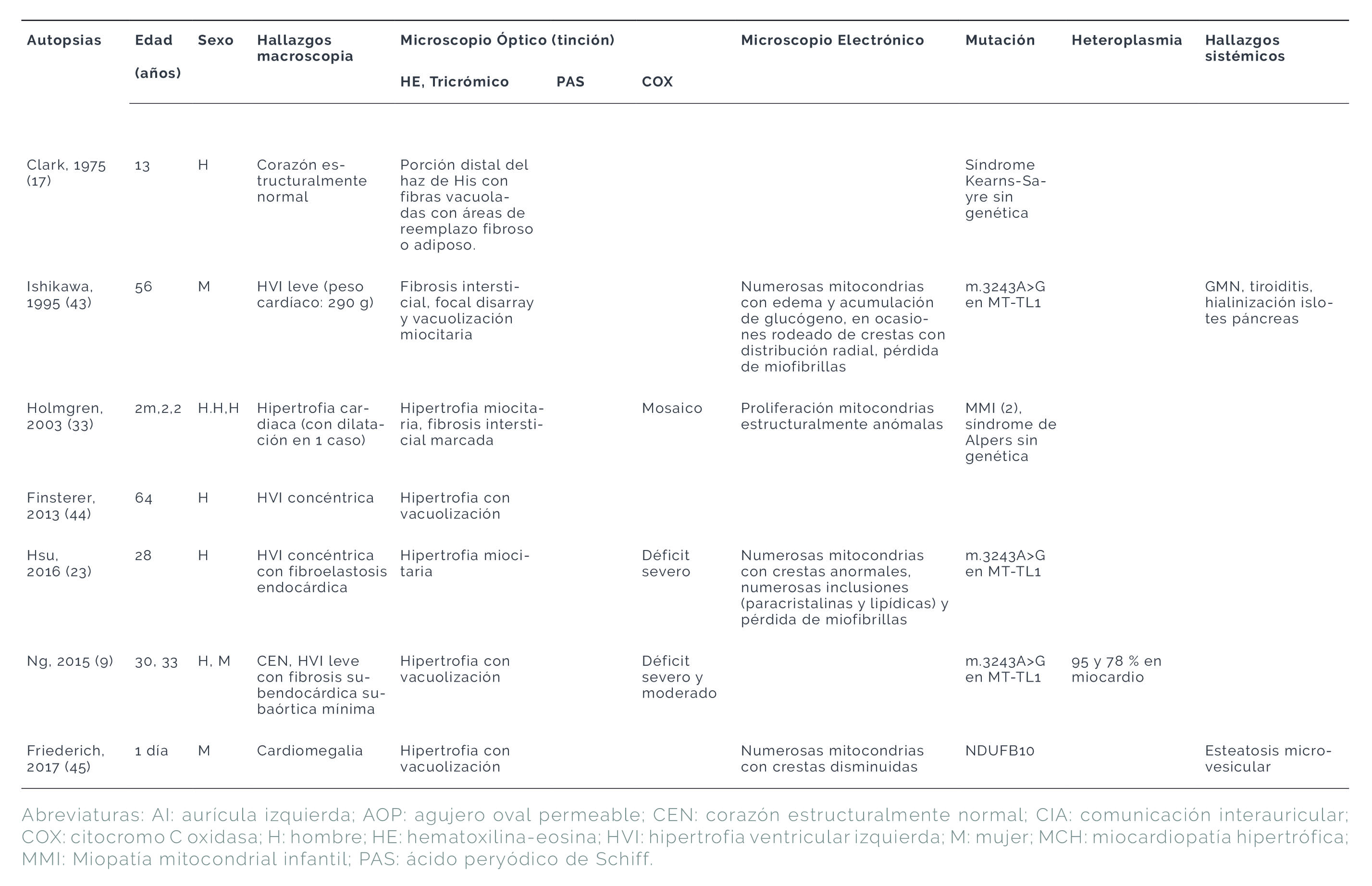
Tabla 2. ter. Hallazgos histopatológicos descritos en los casos de cardiopatías mitocondriales. Casos derivados de autopsias
MICROSCOPÍA ÓPTICA:
- Hipertrofia miocitaria con vacuolización citoplasmática PAS negativa y oil-red positiva (solo realizada en un caso (12)).
- Material granular citoplasmático (mitocondrias) PAS positivo y negativo con PAS-diastasa (confirma glucógeno).
- Fibras rasgadas con tricrómico modificado de Gomori (ver explicación detallada más adelante).
- Fibrosis perimisial y focos de fibrosis de reemplazo (cicatricial).
- Focos de disarray aislados.
- Porción distal del haz de His con fibras vacuoladas de tamaño variable y áreas de reemplazo fibroso o adiposo (en un caso de KSS) (17).
- Déficit variable de COX en miocardiocitos (ver más adelante).
- Inmunohistoquímica MTCO2 positiva (Novus Biologicals), subunidad COX I negativa y subunidad IV positiva (Mitoscience) (18).
MICROSCOPÍA ELECTRÓNICA:
- Acumulación de mitocondrias entre las miofibrillas con desestructuración sarcomérica.
- Alteraciones mitocondriales con diferentes tamaños y formas: gigantes, en forma de anillo, lisis central, crestas con anormal laminación circular concéntrica o con lisis de las crestas en su porción más interna, inclusiones citosólicas cristalinas, paracristalinas, de glucógeno o lípidos, edema y acumulación de glucógeno, en ocasiones rodeado de crestas con distribución radial.
BIOPSIA MUSCULAR
- En clínica, la evaluación en el laboratorio ante la sospecha de una enfermedad mitocondrial es un proceso complejo, en la mayoría de los casos la biopsia muscular constituye el gold standard, incluso cuando no hay evidencia clínica de miopatía. Se utiliza tanto para el estudio histológico como para el oxidativo o el análisis molecular (19).
- El examen histológico e histoquímico del músculo puede ser de gran utilidad para evidenciar la patología mitocondrial. La morfología muscular básica se estudia con las tinciones de hematoxilina-eosina y tricrómico de Gomori modificado.
- En el tricrómico de Gomori modificado, la tinción roja de las mitocondrias se debe a que uno de los ingredientes del colorante reacciona con fosfolípidos abundantes de la membrana mitocondrial (19). Esta tinción permite la detección de fibras de color rojo irregulares denominadas fibras rasgadas. Se caracterizan por una apariencia de “agrietamiento de las fibras” y una proliferación subsarcolémica anormal de las mitocondrias, como resultado de una respuesta compensadora a un defecto bioquímico de la cadena respiratoria. Representan un rasgo histopatológico característico de las EM, sin embargo, no es del todo patognomónico, ya que también se observa con el envejecimiento normal y otras afecciones musculares (5). Aunque en algún caso se pueden observar actividades enzimáticas oxidativas normales (con la mutación m.3243A>G del gen MT-TL1 o algunas mutaciones esporádicas MTCYB, por ejemplo), la mayoría de los pacientes muestran deficiencia de COX asociada a una amplia gama de mutaciones del ADNmt (5). Las fibras rasgadas se encuentran principalmente en KSS, MERRF y MELAS (19).
- Histoquímica enzimática:
- La citocromo C oxidasa (COX) tiñe de ocre el complejo IV, por lo que la tinción estará ausente en presencia de mutaciones en el ADNmt que es responsable de codificar parcialmente esta enzima.
- La succinato deshidrogenasa (SDH) tiñe de azul el complejo II, completamente codificado en el ADNn, por tanto se tiñen normalmente tanto las mitocondrias sanas sin mutación como las de pacientes con mutaciones del ADNmt (19).
- La histoquímica secuencial de COX/SDH es el método estándar utilizado para evaluar la función de la cadena respiratoria mitocondrial en criosecciones musculares. Combinando ambas reacciones, las fibras con disfunción mitocondrial son fácilmente identificables y se ven como una reducción o pérdida en mosaico de la actividad COX con una actividad SDH conservada, indicativo de una anomalía subyacente relacionada con el ADNmt (5).
- La valoración conjunta de SDH y COX tanto en las fibras rojo rasgadas como en las fibras no alteradas permiten, por tanto, orientar el diagnóstico hacia un tipo de enfermedad mitocondrial u otro (19).
- Las fibras rasgadas COX negativas se observan en MERRF, KSS y CPEO las fibras rasgadas COX positivas se observan en MELAS o en mutaciones de genes estructurales mitocondriales (20).
- Microscopía electrónica: normalmente las mitocondrias en el músculo estriado son muy grandes y numerosas y se sitúan debajo del sarcolema, por lo que las alteraciones estructurales de estos organelos se observan fácilmente con el microscopio electrónico. En EM se observan acúmulos de mitocondrias de tamaños diversos, multiformes y en ocasiones con inclusiones en localización subsarcolémica en el músculo estriado.
2. MUERTE SÚBITA ASOCIADA A ENFERMEDAD MITOCONDRIAL
2.1. MUTACIÓN m.3243A>G EN EL GEN MT-TL1
La mutación m.3243A>G en el gen MT-TL1, típica del síndrome MELAS, provoca una reducción de los niveles de síntesis proteica y actividad reducida del complejo I, pero también puede extenderse al complejo IV en casos severos. Esto provoca la reducción de producción de ATP, reducción del potencial de membrana mitocondrial e incremento de la producción de lactato (21).
Majamaa-Voltti y col (2008) (22) identificaron 12 casos de MSC (26%) en un estudio retrospectivo sobre la causa de muerte en once familias (278 personas) con la mutación m.3243A>G en el gen MT-TL1. Aunque no especifican el porcentaje, en estos casos había parientes maternos de primer grado sin afectación clínica. En 5 casos fueron MSC durante el sueño. Además de las enfermedades cardiovasculares, varios de estos pacientes tenían diabetes mellitus y/o epilepsia.
Ng y col (2015) (9) describen dos casos de MSC en dos pacientes con la mutación m.3243A>G en el gen MT-TL1.
- Caso 1: Hombre de 30 años asintomático con dificultad auditiva leve, sin alteraciones en las exploraciones cardiológicas repetidas realizadas en vida y con niveles de heteroplasmia en vida del 92% en orina y 39% en sangre. Falleció durmiendo después de salir con amigos la noche anterior y consumir 10 unidades de alcohol. Su madre había sido diagnosticada con MELAS a los 54 años. Los hallazgos de autopsia fueron normales, compatibles con SMSA. El estudio histoquímico post mortem reveló entre un 40% y un 60% de déficit de COX en ambos ventrículos, con niveles de heteroplasmia del 91% en ventrículo izquierdo, 95% en ventrículo derecho, 92% en tabique interventricular y 93% en ambas aurículas.
- Caso 2: Mujer de 33 años asintomática con leve hipertrofia ventricular izquierda (HVI) diagnosticada en vida, con niveles de heteroplasmia en vida del 68% en orina y 30% en sangre. Falleció durmiendo después de salir con amigos la noche anterior. Su hermana había sido diagnosticada de MELAS con altos niveles de heteroplasmia, siendo su madre y otra hermana también portadoras asintomáticas de la mutación. Los hallazgos de autopsia mostraron leve hipertrofia ventricular izquierda (HVI) y fibrosis subendocárdica mínima en el tracto de salida del ventrículo izquierdo con vacuolización citoplasmática irregular pero prominente e hipertrofia miocitaria. El estudio histoquímico post mortem reveló entre un 15% y un 20% de déficit de COX en ambos ventrículos y unos niveles de heteroplasmia del 76% en ventrículo izquierdo y 78% en ventrículo derecho.
Dadas las circunstancias y la ausencia de pródromos, la causa probable de MSC en estos dos pacientes fue una arritmia maligna cardíaca. Se han descrito frecuentemente arritmias cardíacas en pacientes con m.3243A>G, incluido el síndrome de Wolff-Parkinson-White, la taquicardia supraventricular, la fibrilación auricular y anomalías de despolarización. Este estudio ha calculado una incidencia de SMSA de 2,4 por 1000 personas-año en pacientes con la mutación m.3243A>G en el gen MT-TL1. Hay que destacar que en pacientes con enfermedades de la cadena respiratoria se ha descrito una alta sensibilidad al alcohol. En ambos casos existe el antecedente de cena con amistades la noche previa, confirmándose el consumo de alcohol solo en el primer caso.
Hsu y col (2016) (23) describen la MSC de un hombre de 28 años diagnosticado de MELAS a los 13 años y portador de la mutación m.3243A>G en el gen MT-TL1. Hasta entonces tenía clínica con trastorno convulsivo, baja estatura, miopatía y MCH que fue progresando a una MCD. La autopsia demostró una HVI con marcada fibroelastosis endocárdica y leve dilatación con fibrosis endocárdica en el ventrículo derecho. Microscópicamente se correspondía con una hipertrofia miocitaria con déficit severo de COX, y a nivel ultraestructural se observaron numerosas mitocondrias con crestas anormales, frecuentes inclusiones (paracristalinas y lipídicas) y pérdida de miofibrillas.
Zorio (2021) (24) y Molina y col (2021) (25) identificaron la mutación m.3243A>G en el gen MT-TL1 en un caso de MSC de una mujer de 30 años con migrañas, hipoacusia neurosensorial y calcificación de ganglios basales cuyo diagnóstico post mortem fue de SMSA. El diagnóstico preciso fue posible gracias al estudio de su hermano de 38 años que presentaba hipoacusia neurosensorial, atrofia cerebral supra/infratentorial, miopatía subclínica identificada con electromiografía y biopsia muscular, así como cardiopatía identificada en la ecocardiografía con HVI y disfunción sistólica y en la biopsia endomiocárdica con marcada vacuolización miocitaria citoplasmática PAS negativa y mitocondrias anormales con lisis de las crestas centrales, cuerpos lamelares aislados y pérdida de sarcómeros. El estudio genético identificó la mutación m.3243A>G en el gen MT-TL1 con un nivel de 89-91% de heteroplasmia en sangre en este hombre de 38 años, también en la mujer víctima de SMSA con 30 años con un nivel de heteroplasmia de 23-33% en sangre y 83% en músculo esquelético y miocardio y en la madre de ambos, cardiológicamente asintomática con hipoacusia neurosensorial y migrañas, con un nivel de heteroplasmia del 12-19% en orina y ausente en sangre. Una vez conocida la mutación familiar y demostrada post mortem, se revisó de nuevo la víscera cardíaca de la probando fallecida de una forma más dirigida, observándose: vacuolización generalizada PAS negativa con predominio de áreas subendocárdicas, más marcada en las fibras de Purkinje, degeneración basófila miocitaria llamativa, focos de disarray y ausencia de fibrosis relevante (ver Figura 1).
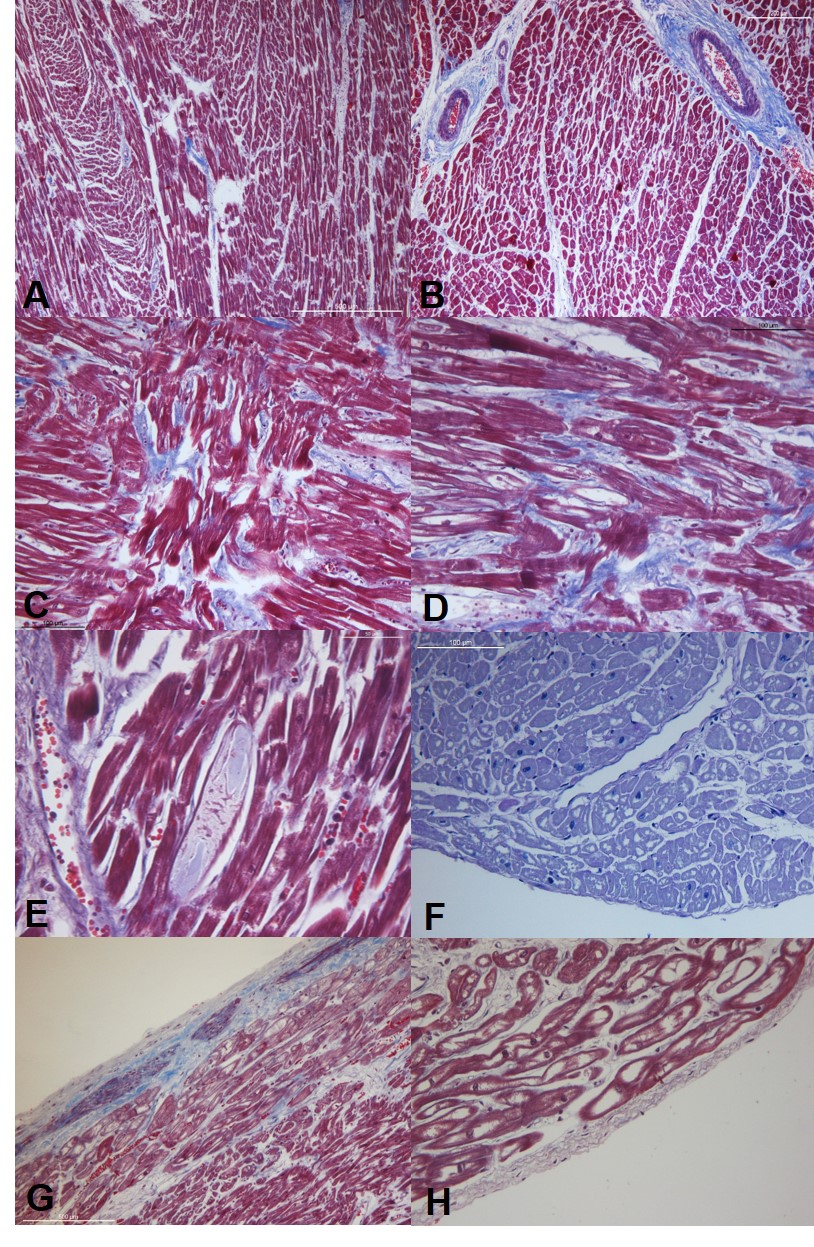
Figura 1. Imágenes de la autopsia de la víctima SMSA portadora de la mutación MT-TL1 m.3243A>G. A: hipertrofia miocárdica con fasciculación y fibrosis perimisial leve; B: enfermedad de pequeño vaso. C y D: focos de disarray con vacuolización miocitaria. E: degeneración basófila miocitaria; F: vacuolización miocárdica negativa en la tinción PAS. G y H: vacuolización miocárdica más prominente en el subendocardio (fibras de Purkinje).
2.2. MUTACIÓN NUCLEAR PPA2
Kennedy y col (2016) (26) identificaron mutaciones bialélicas de la pirofosfatasa inorgánica mitocondrial (PPA2) en 8 individuos (2 adultos) de cuatro familias no relacionadas entre sí que fallecieron de forma súbita, en el marco de un estudio multicéntrico con secuenciación del exoma completo en casos con sospecha de enfermedad mitocondrial.
En la primera familia, se identificó una mutación en dos hermanos que fallecieron de MSC después de consumir pequeñas dosis de alcohol en la segunda década de vida. Ambos habían dado signos de sensibilidad extrema al alcohol previo al acontecimiento. En el examen post mortem se encontró una leve dilatación de ambos ventrículos en uno de los hermanos y en el otro un corazón aumentado de peso y una dilatación del ventrículo izquierdo con fibrosis en forma de lámina circunferencial de tejido cicatricial intramiocárdica. A los otros dos hermanos vivos de esta familia, portadores de la misma mutación en heterocigosis, se les hizo una resonancia magnética cardíaca que demostró una fibrosis miocárdica similar en ambos. A raíz de este hallazgo se les implantó un desfibrilador como prevención primaria de MSC.
Guimier y col (2021) (27) publicaron recientemente un estudio de colaboración internacional en el que reportaron mutaciones del gen PPA2 en 34 casos de 20 familias con clínica de MSC o fallo cardíaco progresivo. En esta serie destaca la alta mortalidad (28/34, 5 casos mayores de 2 años), la presencia de fibrosis miocárdica (en autopsia o resonancia magnética cardíaca) en casi la mitad de los casos (15) con inflamación asociada en 6 de ellos, y, por último, las infecciones víricas (14) y el alcohol (4) como triggers.
2.3. CPEO
En 1965, Kearns (28) realizó una revisión de los antecedentes históricos de casos de oftalmoplejía externa aislada o en asociación con degeneración pigmentaria de la retina o miocardiopatía. De los nueve casos, dos presentaron muerte súbita de probable origen cardíaco (17 y 13 años). Ambos tenían una clínica anterior similar con oftalmoplejía, degeneración pigmentaria de la retina, ptosis, sordera progresiva, miopatía y estatura baja. En uno de los casos se realizó autopsia que evidenció cardiomegalia con HVI y fibrosis subendocárdica.
2.4. KSS
Clark y col (1975) (17) describieron la MSC en un niño de 13 años, diagnosticado desde los 10 años de KSS por ptosis bilateral, miopatía proximal y oftalmoplejía externa. Durante los siguientes tres años, permaneció estable hasta que experimentó un episodio de mareo y síncope por el que fue hospitalizado. Se le detectó un bloqueo completo de la rama derecha, hemibloqueo anterior izquierdo y retraso en la conducción en el haz de His distal o en la división posterior de la rama izquierda. Debido al síncope y los trastornos de conducción cardíaca se implantó un marcapasos y mejoró clínicamente hasta su fallecimiento por MSC cinco meses después. En la autopsia se describió un corazón estructuralmente normal.
El examen microscópico no mostró anomalías del miocardio contráctil, sin embargo, se observaron grandes cambios en el sistema de conducción con hipertrofia y vacuolización de las fibras de Purkinje y fibrosis e infiltración adiposa sin inflamación.
Katsanos y col (2002) (29) reportaron el caso de una mujer de 18 años diagnosticada de KSS que falleció durante un ingreso por una infección de vías respiratorias superiores. Este caso representa el primer paradigma publicado de KSS con daño renal, prolapso de la válvula mitral y defectos de conducción cardíaca.
DISCUSIÓN
Se han encontrado 27 casos bien documentados de MSC en EM (Tabla 3) (9,17,23–29,30,31,32,33,34). No hay predominio de sexo, aunque en 12 casos no consta el dato (22) y el resto corresponden a 7 mujeres y 8 hombres. Todos los casos se encuadran en MSC en menores de 35 años, con una media de edad de 19 años.
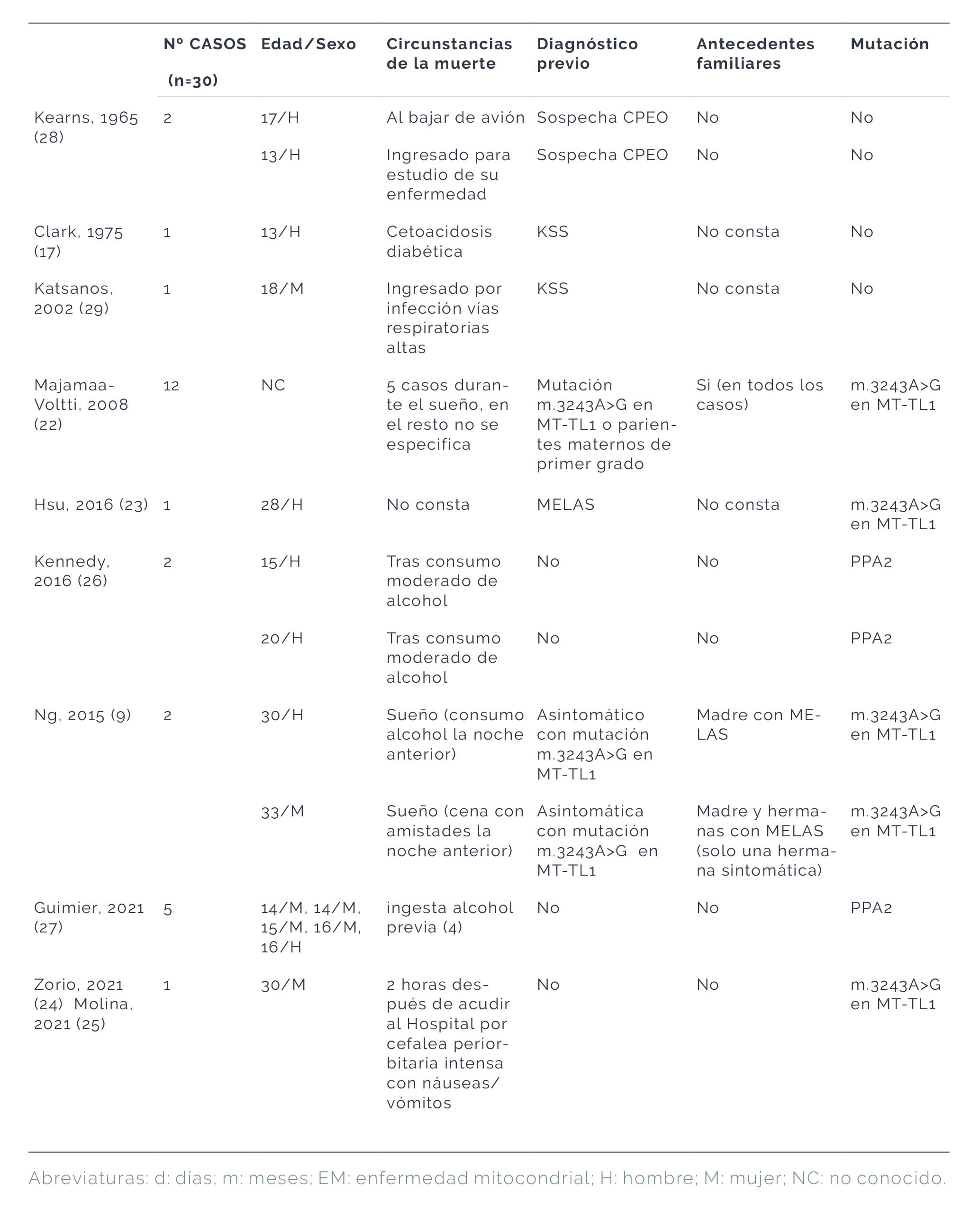
Tabla 3. Casos publicados de MSC en enfermedades mitocondriales.
Aproximadamente la mitad de los casos donde el dato es conocido (15 casos) no tenían un diagnóstico previo (8 casos, 53%) (25,25–27), en 2 casos (11%) había una sospecha clínica ante mortem (27,28) y únicamente en 5 casos (28%) el antecedente de EM era conocido en vida (9,17,23,29), aunque la mayoría eran asintomáticos (53%). La edad media de cada uno de estos grupos varía, siendo mayor en los casos diagnosticados (24 años) en contraposición con los no diagnosticados (17 años) o los dos casos con solo sospecha de EM (15 años).
Las causas de muerte más frecuentes en las EM están relacionadas con la severidad de la afectación clínica, pero existe la posibilidad de que se produzca una MSC en pacientes con clínica leve o asintomáticos, diagnosticados en vida o post mortem. Aunque hay muy pocos casos documentados, la falta de rasgos fenotípicos cardíacos marcados apunta la posibilidad de que exista un infradiagnóstico post mortem.
El estudio genético que confirma el diagnóstico es un dato constante en los trabajos, excepto en los más antiguos (16,17,28,29,33). En adultos, la mutación más identificada es la m.3243A>G en el gen MT-TL1 (MELAS). En una revisión sistemática de afectación cardíaca en miopatías mitocondriales (35) se detectó que los pacientes con deleciones amplias de ADNmt y la mutación m.3243A>G (MELAS) tuvieron fenotipos cardíacos más severos. No obstante, hay casos descritos de portadores de la mutación m.3243A>G del gen MT-TL1 (MELAS) prácticamente asintomáticos (9,22,24,25) sin diagnóstico ante mortem (22). Este dato demuestra la importancia de pensar en esta posibilidad diagnóstica y obtener la máxima información posible en las entrevistas familiares que se realizan en los levantamientos de cadáver (más complicado) y en las Unidades de Cardiopatías Familiares (ambiente más favorable y apropiado) a fin de buscar rasgos característicos (red flags) de EM que refuercen nuestra sospecha diagnóstica (24,25).
De esta revisión se desprende la relación temporal entre algunas de las MSC y el consumo previo de alcohol, constatado en 8 de los 27 casos (30%) (26,27). Esta apreciación coincide con el comentario de Ng y col (2015) (9) afirmando que hay pacientes con defectos en la cadena respiratoria que tienen una alta sensibilidad al alcohol. La base fisiopatológica de esta asociación como trigger de una arritmia cardíaca letal podría radicar en un amento del ácido láctico por dos vías, la debida al defecto en la cadena respiratoria y la secundaria al consumo de alcohol.
La información anatomopatológica en las miocardiopatías mitocondriales procede de las biopsias endomiocárdicas (33,36,37,38,39,40,41,42), de explantes (12,16,18,33) y de estudios autópsicos (9,17,23,33,43,44,45) realizados en casos de EM con diagnóstico clínico y/o genético concluyente. Los rasgos macroscópicos cardíacos identificados en los pacientes vivos mediante técnicas de imagen coinciden con los constatados en las autopsias. Así, en general el rasgo más frecuente es la HVI, con diferentes grados de severidad y con dilatación en algunos casos, pero las manifestaciones clínicas son muy variables. En la Tabla 3 se evidencia cómo existen fenotipos comunes en la mayoría de las publicaciones y rasgos que solo aparecen de forma aislada.
Además de la heterogeneidad en fenotipos macroscópicos con cardiopatía estructural, es cada vez más evidente la posibilidad de encontrar en la autopsia un corazón estructuralmente normal (9,15–17). Ante estos casos, es de crucial importancia que se tenga en cuenta la posibilidad de una EM que haya cursado con una muerte arrítmica, para poder buscar cambios a nivel ultraestructural y realizar los estudios genéticos correspondientes que conduzcan a un correcto diagnóstico.
Respecto a las características microscópicas, los estudios de biopsias endomiocárdicas y explantes cardíacos son más completos técnicamente que en las autopsias, ya que la mayoría incluyen histoquímica y microscopía electrónica. Sin embargo, los estudios post mortem aportan información adicional muy interesante al detectar una mayor afectación de las fibras del sistema de conducción que del miocardio contráctil. Este dato coincide con la posibilidad de que la marcada vacuolización observada en las biopsias endomiocárdicas sea debida a la representación preferencial de tejido subendocárdico en el que se localizan habitualmente las propias fibras de Purkinje, que suelen ser las más afectadas en este escenario clínico. En los trabajos que incluyen imágenes histológicas, las fibras con vacuolización más severa están localizadas a dicho nivel y se disponen en sentido paralelo a la superficie endocárdica con un tamaño ligeramente superior al miocardio contráctil adyacente.
En esta revisión destaca la poca iconografía microscópica óptica encontrada y la diferente metodología aplicada en los diferentes trabajos, con ausencia de una batería diagnóstica homogénea reproducible. El dato más constante y concluyente es la microscopía electrónica, ya que es la clave para sospechar una miocardiopatía mitocondrial. En los estudios post mortem es de menor calidad y falta con frecuencia por la tipología de los tejidos recogidos de forma rutinaria en las autopsias.
La presencia de fibras rasgadas con la tinción tricrómico de Gomori no es habitual encontrarla en el miocardio, pero si es más común en el músculo esquelético, aun sin miopatía evidente (41), por lo que su detección nos podría dar la clave diagnóstica. Por otro lado, la histoquímica con COX da mucha información, pero en el miocardio se realiza en muy pocos casos, posiblemente debido a la necesidad de tener tejido congelado para su realización (9,12,18,23,33,37,38).
Hay también una falta de caracterización de las vacuolas sarcoplásmicas, solo en tres trabajos se demuestra su PAS negatividad (18,40,42) y un único estudio muestra que son positivas con oil red, apuntando una naturaleza lipídica (12). En este último trabajo, en la imagen con la técnica de PAS parece más probable que este material puedan ser mitocondrias aumentadas en número y tamaño (18).
CONCLUSIONES
La MSC asociada a EM es muy infrecuente, pero posiblemente esté infradiagnosticada, ya que requiere un alto nivel de sospecha y un estudio multidisciplinar post mortem con estudios anatomopatológicos y cardiológicos especializados.
La aproximación diagnóstica de una EM en los casos de MSC sin diagnóstico previo deberá basarse en una sintomatología personal y familiar sugestiva (sordera, DM, epilepsia, etc.) y en la presencia de vacuolización miocitaria, con mayor afectación de las fibras de Purkinje. Un segundo paso, que dependerá de la toma de muestras que se haya realizado en el momento de la autopsia, incluiría las técnicas histoquímicas enzimáticas y la microscopía electrónica en miocardio y músculo esquelético. Finalmente, ante la presencia de proliferación mitocondrial anómala, mosaicismo COX y fibras rasgadas será preceptivo el estudio genético confirmatorio.
Es clave sospechar la presencia de una EM para poder solicitar el estudio genético adecuado que lo confirme. Los genes mitocondriales nucleares no se estudian de rutina en estudios genéticos solicitados por cardiólogos ante fenotipos de MCH o MCD y, en caso de tratarse de una EM por mutación en ADNmt, deberá solicitarse de forma específica porque los estudios convencionales sólo se realizan en ADNn y, por tanto, nunca podrán estudiar genes localizados en el ADNmt.
Sería recomendable incluir en los protocolos de autopsia la toma de músculo esquelético (deltoides y/o cuádriceps, por ejemplo) en formol y en congelación (si se dispone de los medios técnicos adecuados) ya que su estudio sistemático básico podría darnos la clave, sin aumentar prácticamente el tiempo y coste de la autopsia.
Se ha asociado una historia de intolerancia al alcohol o ingesta ante mortem. Son necesarios más estudios sobre la acción del alcohol en los mecanismos etiopatogénicos de la MSC por EM. Sin embargo, debido a esta asociación observada en varios casos, la toma de alcohol, por pequeña que sea, podría actuar como trigger en estos pacientes y considerarse un signo de alarma/sospecha (red flag) de EM.
Consideramos de suma importancia mejorar el conocimiento en EM de los especialistas relacionados para aumentar su sensibilidad diagnóstica ante estas enfermedades. Hoy en día, el bajo nivel de formación en estas enfermedades, su enorme variabilidad clínica y las destacables dificultades diagnósticas dependientes de pruebas invasivas (biopsias) y estudios no rutinarios (como es el estudio de ADNmt) genera grandes trabas para diagnosticar correctamente a estos pacientes. Es necesario entender mejor cómo las EM afectan al corazón para poder desarrollar los algoritmos terapéuticos ajustados a la enfermedad subyacente, tal y como propugna la tan deseada Medicina Personalizada. Si en el actual trabajo hemos comprobado que la MS puede producirse, de forma impredecible, con niveles bajos (20-30%) de heteroplasmia en sangre (que suele ser el biofluido más accesible en la práctica clínica diaria), resulta todo un reto avanzar en la lucha contra la MSC en este escenario con los conocimientos disponibles en estos momentos. Estamos a tiempo de mejorar esta realidad y el presente trabajo pretende aportar un grano de arena en este camino.
Conflicto de Intereses
Los autores/as de este artículo declaran no tener ningún tipo de conflicto de intereses respecto a lo expuesto en el presente trabajo.
FUENTES DE FINANCIACIÓN
Ninguna.
BIBLIOGRAFÍA
- ↑ Priori SG, Blomström-Lundqvist C, Mazzanti A, Blom N, Borggrefe M, Camm J, et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC)Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur Heart J. 2015;36(41):2793-867.
- ↑ Tashiro, R. , Onoue, N. , Shinozaki, T. . Mitochondrial Cardiomyopathy. In: Tsipis, A. , editor. Current Perspectives on Cardiomyopathies [Internet]. London: IntechOpen; 2018 [cited 2022 Jun 07]. Available from: https://www.intechopen.com/chapters/61880 doi: 10.5772/intechopen.77105
- ↑ Mas LG. El auge de las enfermedades mitocondriales. SEQC. 2018;11.
- ↑ Feillet F, Schmitt E, Gherardi R, Bonnemains C. Enfermedades mitocondriales. EMC – Pediatría. 2014;49(2):1-12.
- ↑ Alston CL, Rocha MC, Lax NZ, Turnbull DM, Taylor RW. The genetics and pathology of mitochondrial disease: Mitochondrial genetic disease. J Pathol. 2017;241(2):236-50.
- ↑ Meyers DE, Basha HI, Koenig MK. Mitochondrial Cardiomyopathy. Tex Heart Inst J. 2013;40(4):10.
- ↑ Leonard J, Schapira A. Mitochondrial respiratory chain disorders I: mitochondrial DNA defects. The Lancet. 2000;355(9200):299-304.
- ↑ Gorman GS, Schaefer AM, Ng Y, Gomez N, Blakely EL, Alston CL, Feeney C, Horvath R, Yu-Wai-Man P, Chinnery PF, Taylor RW, Turnbull DM, McFarland R. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann Neurol. 2015 May;77(5):753-9
- ↑ Ng YS, Grady JP, Lax NZ, Bourke JP, Alston CL, Hardy SA, et al. Sudden adult death syndrome in m.3243A>G-related mitochondrial disease: an unrecognized clinical entity in young, asymptomatic adults. Eur Heart J. 2015;37(32):2552-9.
- ↑ Chinnery PF. Mitochondrial disease in adults: what’s old and what’s new? EMBO Mol Med. 2015;7(12):1503-12.
- ↑ Mazzaccara C, Mirra B, Barretta F, Caiazza M, Lombardo B, Scudiero O, et al. Molecular Epidemiology of Mitochondrial Cardiomyopathy: A Search Among Mitochondrial and Nuclear Genes. Int J Mol Sci. 2021;22(11):5742.
- ↑ Bates MGD, Bourke JP, Giordano C, d’Amati G, Turnbull DM, Taylor RW. Cardiac involvement in mitochondrial DNA disease: clinical spectrum, diagnosis, and management. Eur Heart J. 2012;33(24):3023-33.
- ↑ Vydt TC, de Coo RF, Soliman OI, Ten Cate FJ, van Geuns RJ, Vletter WB, Schoonderwoerd K, van den Bosch BJ, Smeets HJ, Geleijnse ML. Cardiac involvement in adults with m.3243A>G MELAS gene mutation. Am J Cardiol. 2007 Jan 15;99(2):264-9.
- ↑ Finsterer J. Histiocytoid Cardiomyopathy: A Mitochondrial Disorder. Clin Cardiol. 2008;31(5):225-7.
- ↑ Kabunga P, Lau AK, Phan K, Puranik R, Liang C, Davis RL, et al. Systematic review of cardiac electrical disease in Kearns–Sayre syndrome and mitochondrial cytopathy. Int J Cardiol. 2015;181:303-10.
- ↑ Schwartzkopff B, Frenzel H, Breithardt G, Deckert M, Lösse B, Toyka KV, et al. Ultrastructural findings in endomyocardial biopsy of patients with Kearns-Sayre syndrome. J Am Coll Cardiol. 1988;12(6):1522-8.
- ↑ Clark DS, Myerburg RJ, Morales AR, Befeler B, Hernandez FA, Gelband H. Heart Block in Kearns-Sayre Syndrome. 1975;68(5):727-30.
- ↑ Giordano C, Perli E, Orlandi M, Pisano A, Tuppen HA, He L, et al. Cardiomyopathies due to homoplasmic mitochondrial tRNA mutations: morphologic and molecular features. Hum Pathol. 2013;44(7):1262-70.
- ↑ Ridaura-Sanz DC. La biopsia en el diagnóstico de la enfermedad pediátrica. Acta Pediatr Mex 2008;29(6):347-54.
- ↑ Garcia Villanueva M. Contribución de la anatomía patológica al diagnóstico de las enfermedades musculares. Mesa redonda: Hipotonía en el neonato y el lactante (I). Comunicación presentada en: IX Curso de enfermedades musculares en la infancia y adolescencia;2012, 22 de marzo; Madrid.
- ↑ Cuadros Arasa M. Efecto de las mutaciones en el ADN mitocondrial sobre la expresión de genes implicados en la función mitocondrial. [tesis doctoral], Barcelona, Universitat Autònoma de Barcelona; 2013. http://hdl.handle.net/10803/113563
- ↑ Majamaa-Voltti K, Turkka J, Kortelainen ML, Huikuri H, Majamaa K. Causes of death in pedigrees with the 3243A>G mutation in mitochondrial DNA. J Neurol Neurosurg Psychiatry. 2008;79(2):209-11.
- ↑ Hsu YHR, Yogasundaram H, Parajuli N, Valtuille L, Sergi C, Oudit GY. MELAS syndrome and cardiomyopathy: linking mitochondrial function to heart failure pathogenesis. Heart Fail Rev. 2016;21(1):103-16.
- ↑ Zorio E. Caso clínico: imagen y cardiopatías familiares. Comunicación presentada en: VII Reunión virtual de la Sección de Cardiopatías Familiares y Genética Cardiovascular de la Sociedad Española de Cardiología;2021, 4-6 de febrero.
- ↑ Molina P, Mancheño N, Muelas N, Azorín I, Zorio E. Caso clínico: muerte súbita. Comunicación presentada en: VII Reunión virtual de la Sección de Cardiopatías Familiares y Genética Cardiovascular de la Sociedad Española de Cardiología;2021, 4-6 de febrero.
- ↑ Kennedy H, Haack TB, Hartill V, Mataković L, Baumgartner ER, Potter H, et al. Sudden Cardiac Death Due to Deficiency of the Mitochondrial Inorganic Pyrophosphatase PPA2. Am J Hum Genet. 2016;99(3):674-82.
- ↑ Guimier A, Achleitner MT, Moreau de Bellaing A, Edwards M, de Pontual L, Mittal K, et al. PPA2-associated sudden cardiac death: extending the clinical and allelic spectrum in 20 new families. Genet Med. 2021;23(12):2415-25.
- ↑ Kearns TP. External ophthalmoplegia, pigmentary degeneration of the retina, and cardiomyopathy: a newly recognized syndrome. TR AM OPHTH Soc. 1965;63:67.
- ↑ Katsanos KH, Pappas CJ, Patsouras D, Michalis LK, Kitsios G, Elisaf M, et al. Alarming atrioventricular block and mitral valve prolapse in the Kearns–Sayre syndrome. Int J Cardiol. 2002;83(2):179-81.
- ↑ Guimier A, Gordon CT, Godard F, Ravenscroft G, Oufadem M, Vasnier C, et al. Biallelic PPA2 Mutations Cause Sudden Unexpected Cardiac Arrest in Infancy. Am J Hum Genet. 2016;99(3):666-73.
- ↑ Vasilescu C, Ojala TH, Brilhante V, Ojanen S, Hinterding HM, Palin E, et al. Genetic Basis of Severe Childhood-Onset Cardiomyopathies. J Am Coll Cardiol. 2018;72(19):2324-38.
- ↑ Phoon CKL, Halvorsen M, Goldstein DB, Rabin R, Cecchin F, Crandall L, Devinsky O. Sudden unexpected death in asymptomatic infants due to PPA2 variants. Mol Genet Genomic Med. 2020 Jan;8(1):e1008.
- ↑ Holmgren D. Cardiomyopathy in children with mitochondrial disease Clinical course and cardiological findings. Eur Heart J. 2003;24(3):280-8.
- ↑ Majamaa-Voltti K, Peuhkurinen K, Kortelainen ML, Hassinen IE, Majamaa K. Cardiac abnormalities in patients with mitochondrial DNA mutation 3243A>G. BMC Cardiovasc Disord. 2002;2(1):12.
- ↑ Quadir A, Pontifex CS, Lee Robertson H, Labos C, Pfeffer G. Systematic review and meta-analysis of cardiac involvement in mitochondrial myopathy. Neurol Genet. 2019;5(4):e339.
- ↑ Nakanishi M, Harada M, Tadamura E, Kotani H, Kawakami R, Kuwahara K, et al. Mitochondrial Cardiomyopathy Evaluated With Cardiac Magnetic Resonance. 2007;116:e25–e26
- ↑ Jose T, Gdynia HJ, Mahrholdt H, Vöhringer M, Klingel K, Kandolf R, et al. CMR gives clue to “ragged red fibers” in the heart in a patient with mitochondrial myopathy. Int J Cardiol. 2011;149(1):e24-7.
- ↑ Palecek T, Tesarova M, Kuchynka P, Dytrych V, Elleder M, Hulkova H, et al. Hypertrophic Cardiomyopathy Due to the Mitochondrial DNA Mutation m.3303C>T Diagnosed in an Adult Male. Int Heart J. 2012;53(6):383-7.
- ↑ Takemura G, Onoue K, Kashimura T, Kanamori H, Okada H, Tsujimoto A, et al. Electron Microscopic Findings Are an Important Aid for Diagnosing Mitochondrial Cardiomyopathy With Mitochondrial DNA Mutation 3243A>G. Circ Heart Fail. 2016;9(7).
- ↑ Houston BA, Judge DP, Brown E, Halushka M, Barouch LA. Giant Ring Mitochondria in a Patient With Heart Failure and Cerebral White Matter Disease Resulting From an MT-TL1 Mitochondrial Gene Mutation. J Card Fail. 2017;23(8):652-5.
- ↑ Kuno T, Imaeda S, Asakawa Y, Nakamura H, Takemura G, Asahara D, et al. Mitochondrial Cardiomyopathy Presenting as Dilated Phase of Hypertrophic Cardiomyopathy Diagnosed with Histological and Genetic Analyses. Case Rep Cardiol. 2017;2017:1-4.
- ↑ Yamasaki T, Yanishi K, Tateishi S, Nakanishi N, Zen K, Nakamura T, et al. Late-onset Mitochondrial Cardiomyopathy Triggered by Anticancer Treatment. Intern Med. 2017;56(11):1357-61.
- ↑ Ishikawa Y, Asuwa N, Ishii T, Masuda S, Kiguchi H, Hirai S, et al. Severe Mitochondrial Cardiomyopathy and Extraneuromuscular Abnormalities in Mitochondrial Encephalomyopathy, Lactic Acidosis, and Stroke-like Episode (MELAS). Pathol – Res Pract. 1995;191(1):64-9.
- ↑ Finsterer J, Stöllberger C, Grassberger M, Gerger D. Noncompaction in Mitochondrial Myopathy: Visible on Microscopy but Absent on Macroscopic Inspection. Cardiology. 2013;125(3):146-9.
- ↑ Friederich MW, Erdogan AJ, Coughlin CR, Elos MT, Jiang H, O’Rourke CP, et al. Mutations in the accessory subunit NDUFB10 result in isolated complex I deficiency and illustrate the critical role of intermembrane space import for complex I holoenzyme assembly. Hum Mol Genet. 2017 Feb 15;26(4):702-716.
- ↑ Ryzhkova A, Sazonova M, Sinyov V, Galitsyna E, Chicheva M, Melnichenko A, et al. Mitochondrial diseases caused by mtDNA mutations: a mini-review. Ther Clin Risk Manag. 2018;Volume 14:1933-42.
- ↑ Leonard J, Schapira A. Mitochondrial respiratory chain disorders II: neurodegenerative disorders and nuclear gene defects. The Lancet. 2000;355(9201):389
Correspondencia:
Esther Zorio Grima
Servicio de Cardiología. Avenida Fernando Abril Martorell, 106 · 46026 Valencia
E-mail: zorio_est@gva.es
Tlf.: 961 245 849