Original
Guerra Civil Española (1936-1939): identificación de restos humanos procedentes de fosas comunes en Cataluña mediante análisis de ADN Mitocondrial. A propósito de un caso.
Spanish Civil War (1936-1939): identification of human remains from mass graves in Catalonia using mitochondrial DNA analysis. A case report.

Cuad Med Forense. 2004; 10(38):37-46
Fecha de recepción: 31.MAR.05; Fecha de aceptación: 17.JUN.05
RESUMEN
La identificación de personas desaparecidas durante la Guerra Civil española se ha convertido en un tema de interés social. El presente trabajo describe la identificación del primer cadáver exhumado en Cataluña procedente de una fosa común de la guerra civil. Como técnicas confirmativas a la metodología clásica empleada en la identificación de restos humanos (antropométricas, fórmula del análisis dental...) la ciencia forense encuentra en el análisis del ADN una herramienta útil. La identificación se llevó a cabo analizando la región de control del ADN mitocondrial debido a la antigüedad de los restos humanos (aproximadamente 65 años) y a que la única muestra de referencia disponible procedía de un lejano pariente materno (sobrina). Los productos de secuenciación fueron analizados utilizando un ABI PRISM 310 (AB). Tras cumplir con todos los requisitos de aceptación de las secuencias y descartada la posible presencia de contaminaciones eventuales, se observó una total coincidencia entre las secuencias del ADNmt del fémur y la muestra de referencia aportada por el familiar. La secuencia obtenida era 16298C, 263G, 315.1C y después de consultar una base de datos de 2192 individuos caucásicos esta secuencia fue observada una vez, de modo que la frecuencia de esta secuencia es de 0.0013. Los resultados obtenidos apoyan el hecho de que cuando los análisis convencionales de ADN nuclear son inviables el análisis de ADNmt se convierte en una útil y poderosa herramienta.
Palabras clave: ADN mitocondrial, PCR, Restos Humanos, Medicina Forense, Guerra Civil Española, Identificación.
ABSTRACT
The identification of missing people during the Spanish civil war has become a subject of social interest. The present work describes the identification of the first corpse from a mass grave exhumed in Catalonia. As confirmatory technique to the classic methodology used in the identification of human skeletal remains (anthropometrics, dental charts...) forensic science finds in the DNA analysis an useful tool. The identification was carried out analyzing the control region of mitochondrial DNA due to the antiquity of the human remains (approximately 65 years) and because the only available sample of reference came from a distant maternal relative (niece). The sequencing products were analyzed using the genetic analyzer ABI PRISM 310 (AB). After fulfilling all the requirements of acceptance of the sequences and eliminated the possible presence of contaminations, a match was observed between the mtDNA sequences obtained for both samples, femur and the sample of reference contributed by the relative. The obtained sequence was 16298C, 263G, 315.1C and only one ocurrence of this sequence was observed in a database of 2192 Caucasian individuals (frequency 0,0013). The obtained results support the fact that when conventional nuclear DNA typing is not possible the analysis of mtDNA becomes an useful and powerful tool.
Key words: Mitochondrial DNA, PCR, Human Remains, Forensic Medicine, Spanish Civil War, Identification.
INTRODUCCIÓN
En los últimos años en España diferentes colectivos sociales representados fundamentalmente por familiares de personas desaparecidas durante la guerra civil revindican lo que se ha venido en llamar «el reconocimiento de la memoria histórica». A esta sensibilidad no ha sido ajeno el Parlamento de Cataluña y en el año 2003 se acordó crear una comisión multidisciplinar de expertos con la intención entre otras cuestiones de realizar la identificación de cadáveres procedentes de fosas comunes ubicadas en Cataluña.
En el presente trabajo se describe la identificación mediante pruebas de análisis genético del primer cadáver exhumado de una fosa común de la Guerra Civil Española (1936-1939) en Cataluña.
En ocasiones, la identificación de cadáveres mediante las técnicas clásicas de la medicina forense (huellas dactilares, fórmula dentaria, medidas antropológicas ) no permiten definir con precisión la identidad de unos restos cadavéricos y es necesario recurrir a técnicas de análisis de ácido desoxirribonucleico (ADN).
En las dos últimas décadas los avances en biología molecular han permitido dotar a la medicina forense con una valiosísima herramienta para la resolución de casos judiciales tanto en el ámbito penal como en el civil. Efectivamente, hoy mediante el análisis de polimorfismos de ADN somos capaces de precisar con altos índices de probabilidad el autor de una violación, un homicidio o la identidad de un cadáver.
Desgraciadamente existen dos factores como son el tiempo transcurrido y la condiciones medioambientales (humedad, temperatura ) que ocasionan una degradación del material genético. Esta circunstancia hace que al quedar afectado el ADN su análisis mediante marcadores genéticos nucleares sea en ocasiones inviable. La medicina forense dispone en la actualidad de otra estrategia para este tipo de muestras, se trata del análisis del ADN ubicado en las mitocondrias y al que conocemos como mitocondrial (ADNmt).
La molécula de ADNmt es una molécula circular y cerrada de 16569 pares de bases. La proporción de las cuatro bases es desigual en ambas hebras, de manera que una de ellas es rica en adeninas y guaninas (hebra pesada ) mientras que la otra es rica en citosinas y timinas (hebra ligera).
La zona del ADNmt más comúnmente empleada con fines forenses es una región caracterizada por una alta variabilidad entre individuos llamada región de control o d-loop. Dentro de dicha zona se encuentran ubicadas dos regiones llamada región hipervariable 1 (HV1) que se extiende entre las bases 16024 y 16365 y la región hipervariable 2 (HV2) comprendida entre las bases 73 y 340 (Fig. 1). La región de control no codifica para proteínas, ARNs de transferencia o RNAs ribosómicos, sin embargo en ella se encuentran ubicados los promotores de las hebras pesada y ligera, sitios de unión para factores de transcripción mitocondrial o el origen de replicación de la hebra pesada.
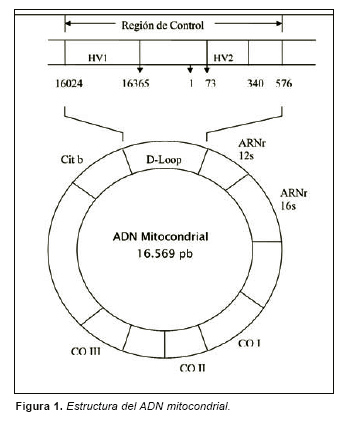
Existen tres características que hacen del ADNmt una herramienta idónea para la resolución de casos como el que aquí se describe. En primer lugar, mientras que el ADN nuclear está presente en dos copias por célula diploide el número de moléculas de ADNmt por célula puede oscilar entre 1000 y 10000 moléculas [1]. Esta circunstancia hace viables análisis genéticos cuando estamos ante material biológico con escasa cantidad de ADN nuclear o ADN parcialmente degradado. En segundo lugar, se ha constatado que en las regiones sometidas a estudio (HV1 y HV2) el índice de mutación es entre 5 y 10 veces superior al que acontece en el ADN nuclear. [2-3]. Por último, el ADNmt es heredado por vía materna [4], de manera que salvo que ocurran procesos de mutación, todas las personas que estén emparentados por vía materna poseerán el mismo ADNmt. Esta propiedad es sumamente importante puesto que nos va a permitir identificaciones de restos cadavéricos incluso habiendo transcurrido varias generaciones [5-6].
MATERIAL Y MÉTODOS:
Todo el proceso de análisis genético fue realizado en el Servicio de Biología del Instituto Nacional de Toxicología y Ciencias Forenses del Departamento de Barcelona.
Organización del laboratorio y aceptación de secuencias.
El gran enemigo que afrontan los laboratorios que practican análisis genéticos con fines forenses se llama contaminación. La aparición de material genético exógeno al propio de la evidencia ocasiona resultados no interpretables y en algunos casos absolutamente erróneos. Esta circunstancia se agudiza especialmente en aquellas muestras en las que la cantidad de ADN presente es muy escasa. Es por tanto muy importante seguir unas medidas orientadas a minimizar la aparición de contaminaciones. Durante todo el proceso de extracción y amplificación se empleó bata, mascarilla y guantes que fueron cambiados con frecuencia. La extracción de muestras, tanto dubitadas (huesos) como indubitadas (saliva y pelos), se llevó a cabo en campanas de flujo laminar; asimismo la extracción de muestras dubitadas e indubitadas se realizó con un intervalo de tiempo y en habitaciones separadas. Reactivos y tubos fueron autoclavados previamente a su uso. Con el fin de chequear reactivos en los pasos de extracción y amplificación se emplearon blancos control. La secuenciación de las regiones HV1 y HV2 se realizó tanto en sentido 5′-3 como en el sentido inverso 3′-5′. Todas las extracciones y amplificaciones se llevaron a cabo por dos personas por separado y de la misma forma se contrastaron los resultados obtenidos, asimismo estos resultados finales fueron cotejados con las secuencias de ADNmt del personal del laboratorio. Hasta que no se dispuso de la secuencia correspondiente al fémur no se solicitó la muestra de refencia para su cotejo, impidiendo de esta manera cualquier posibilidad de cambio de muestras en el laboratorio.
Las secuencias finales se aceptaron cuando se cumplían todos los requisitos que a continuación describimos:
1.Blancos control de extracción y amplificación eran negativos.
2. Lecturas de secuenciación, para cada una de las regiones, coincidentes en sentido 5′-3′ y 3′-5′.
3. Resultados finales de secuencia coincidente para los extractos independientes realizados por dos operarios.
4. No coincidencia de los resultados finales con las secuencias del personal del laboratorio.
Muestras.
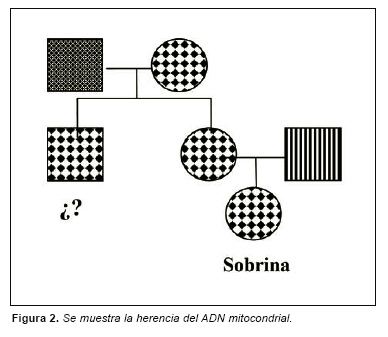
A tenor de la información proporcionada por gente del lugar en la fosa que se exhumó en Prat de Lluçanes (Osona-Cataluña-), cabía esperar la aparición de seis soldados republicanos y un civil. El trabajo antropológico sobre los restos óseos aparecidos permitió seleccionar que cadáver podría corresponder con el civil, siendo muestras de este cadáver las que fueron remitidas al laboratorio para confirmar su identidad. Se remitió un fémur derecho y otro izquierdo de aproximadamente 51cm de longitud del cadáver que se presumía podía ser el civil. Una vez recibidas estas muestras se almacenaron a -20 ºC hasta su análisis. Con posterioridad se proporcionó muestras de referencia consistente en saliva y pelos con raíz procedentes de un presunto familiar, concretamente una sobrina por línea materna de la persona a la que presumiblemente se esperaba que perteneciesen los huesos (Fig. 2).
Extracción de material genético.
Para obtener el ADN a partir de las muestras indubitadas de saliva y pelo se empleó un método clásico de extracción mediante disolventes orgánicos y posterior concentración con Microcon-100 (Millipore, Billerica, MA, USA).
Se emplearon aproximadamente 2 gr de la porción central de fémur para la extracción de ADN. Con el fin de eliminar todo el posible material contaminante que el hueso pudiera presentar en su capa más externa se limpió este con papel abrasivo de lija. Tras este paso se recuperaron 2 gr de fino polvo de hueso con la ayuda de una lima que se depositaron en tubos de 50 mL con 8 mL de EDTA 0,5 M pH 7,5 , 1 mg/ml de proteinasa K (10 mg/mL) y 6 % de sodio dodecil sulfato (SDS).
Los tubos se pusieron en agitación a 56 ºC durante 18 h. El polvo de hueso no digerido fue sedimentado por centrifugación y el sobrenadante fue decantado a otro tubo de 50 mL. El extracto fue limpiado de proteínas con el uso de fenol:cloroformo:isoamílico (25:24:1) y posteriormente el ADN se concentró y liberó de posibles inhibidores mediante ultrafiltración en columnas Centricon-100 (Millipore, Billerica, MA, USA).
Cuantificación de ADN.
La calidad del ADN extraído se estimó mediante el empleo de electroforesis submarina en minigeles de agarosa y posterior tinción con bromuro de etidio.
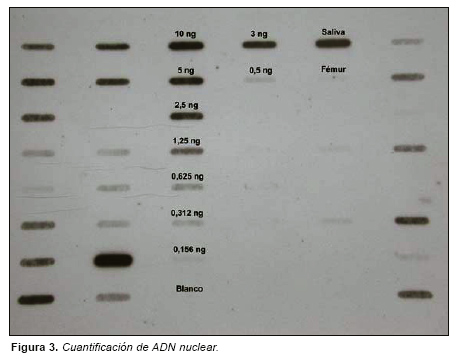
La cantidad de ADN humano procedentes tanto de las muestra de fémur como de las muestras indubitadas de saliva y pelo se cuantificó mediante hibridación con la sonda D17Z1 primate específica utilizando el kit Quantiblot (Applied Biosystems, Foster City, USA). (Fig 3).
Amplificación y secuenciación.
Las zonas sometidas a análisis fueron las regiones HV1 comprendida entre los nucleotidos 16024 y 16365 y la región HV2 ubicada entre las bases 73 y 340. La numeración de las bases se hace en función a la asignación realizada en la primera secuenciación del ADNmt por Anderson y cols [7] que sirve como secuencia de referencia en el campo forense y es conocida como CRS (Cambridge Reference Sequence). Los primers empleados en la amplificación de ambas regiones son los descritos por Wilson y cols [8]. La región HV1 fue amplificada con la pareja de primers L15997 y H16395 mientras que para la región HV2 se emplearon los primers L048 y H408. La tabla 1 describe la secuencia de cada uno de los primers empleados. La reacción de amplificación se llevó a cabo en un termociclador GeneAmpPCR System 2400 (Applied Biosystems, Foster City, USA). Las amplificaciones de cada una de las regiones se llevó a cabo por separado y cada reacción de amplificación contenía 5 pmol de cada primer, 50 mM de KCl, 10 mM Tris-HCl (pH 8.3), 1.5 mM de MgCl2, 0,001% de gelatina, 200 µM de cada nucleótido, 5U de AmpliTaqGold polimerasa (Applied Biosystems, Foster City, USA).

Los extractos de ADN procedentes de las muestras de hueso fueron amplificados en un volumen final de 50 µl y con la adición de 0,2 mg/ml de BSA (suero de albúmina bovina) para minimizar la acción de inhibidores de la PCR y que presumiblemente cabe esperar en muestras de la antigüedad de estas (65 años). Se utilizaron para la amplificación entre 1 y 3 µl de extracto mientras que de las muestras indubitadas bastó con 1µ con un volumen final de 25 µl, no teniéndose que emplear BSA.
Con el fin de minimizar el riesgo de contaminación se amplificaron por separado las muestras dubitadas e indubitadas. La no presencia de ADN contaminante en los reactivos empleados en la amplificación se chequeó mediante la amplificación de un blanco de reactivos.
La eficiencia de la amplificación se evaluó mediante electroforesis submarina en minigeles de agarosa y posterior tinción con bromuro de etidio. Los productos de amplificación tanto de las muestras problema como de los controles negativos de extracción y amplificación se compararon con patrones de concentración y longitud conocidos (Fig 4).

El material amplificado se purificó de nucleótidos y primers no incorporados mediante ultrafiltración en columnas Centricon-100 (Millipore, Billerica, MA, USA).
Se emplearon 3 µl de producto de PCR purificado para llevar a cabo la reacción de secuenciación para la que se utilizó el kit BigDye™ Cycle Sequencing (Applied Biosystems, Foster City, USA), siguiendo las instrucciones del suministrador. La posterior eliminación de terminadores no incorporados se consiguió mediante el empleo de columnas Centri-Sep™ (Princepton Separations Inc).
La lectura de secuencias fue llevada a cabo en un secuenciador automático ABI PRISM™ 310 (Applied Biosystems, Foster City, USA) y para la posterior edición e interpretación de resultados se empleó el software SeqScape™ desarrollado por Applied Biosystems (versión 2.1.1).
Evaluación de los resultados.
En aquellos casos en los que no se puede excluir que las muestras dubitadas e indubitadas puedan proceder de la misma línea materna es conveniente aportar algún parámetro estadístico que nos de idea del peso de dicha coincidencia. Tal y como recoge la Internacional Society of Forensic Genetics (ISFG ) [9] y el European DNA profiling group (EDNAP) [10] en sendas publicaciones, lo aconsejable para poder calcular la probabilidad (p) con la que se presenta una determinada secuencia es realizar un contaje del número de veces que esa secuencia aparece en una base de datos y posteriormente aplicarle un factor de corrección por error de muestreo tales como un cálculo del intervalo de confianza [11] o la corrección de Balding y Nichols [12]. Estos factores de corrección son necesarios puesto que en las bases de datos de las que se dispone en la actualidad es relativamente escaso el número de secuencias y es de esperar que vayan apareciendo con cierta frecuencia variantes que no se encuentran aún en ellas.
En el presente trabajo las secuencias se evaluaron empleando el factor de corrección de Balding y Nichols:
p = (x + 2) / (n + 2)
Donde x es el número de observaciones de la secuencia obtenida en una base de datos de n individuos.
Asimismo, una vez conocida la probabilidad con la que aparece una determinada secuencia en una base de datos, según recomienda la ISFG, lo deseable es realizar una aproximación bayesiana de dicha coincidencia entre muestra dubitada e indubitada, y para ello se enfrentan hipótesis alternativas. Se emplea para ello el índice de verosimilitud o «likelihood ratio» (LR) [13].
RESULTADOS Y DISCUSIÓN:
La cuantificación de las muestras de hueso sometidas a análisis puso de manifiesto la presencia de cantidades de ADN nuclear ligeramente inferiores a 156 pgr (Fig. 3).
El análisis del minigel de los productos amplificados de los extractos de ADN de fémur (Fig. 4) nos indicó que, a pesar de observarse un alto grado de degradación en dichas muestras, el resultado de la reacción de amplificación fue eficiente para fragmentos de aproximadamente 340 pb (HV1) y 290 pb (HV2). Previa a esta eficiente amplificación se había realizado una primera amplificación en un volumen final de 25 µl y sin emplear BSA, no obteniéndose resultado alguno. Se comprobó que la causa de esta infructuosa reacción de amplificación era achacable probablemente a la presencia de inhibidores en el extracto de ADN, puesto que al incorporar en el mismo tubo de amplificación el mismo volumen de ADN empleado en la amplificación cuyo resultado había sido negativo y un control positivo el resultado final de la amplificación también fue negativo. Con el fin de diluir los inhibidores se aumentó el volumen del tubo de la reacción de PCR a 50 µl y se incorporó una sustancia que facilita la reacción de PCR como es el BSA, obteniéndose así resultados positivos en este caso.
Las muestras indubitadas mostraron resultados de amplificación ampliamente satisfactorios sin tener que recurrir a empleo de un doble volumen de amplificación ni BSA. Quedó garantizada la ausencia de ADN contaminante en las reacciones de extracción y amplificación por los resultados negativos en la amplificación de los blancos de extracción y amplificación.
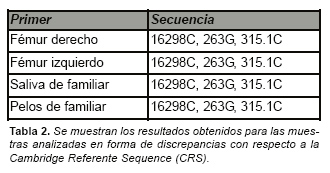
La secuencia de nucleótidos obtenida para las regiones HV1 y HV2 del ADNmt fue completamente coincidente entre las dos extracciones realizadas sobre los huesos (fémur derecho y fémur izquierdo). En la región HV1 presentaba una discrepancia con respecto a la CRS en la posición 16298, concretamente una transición de una timina a citosina. La región HV2 presentó dos discrepancias sumamente usuales, por una parte una transición de adenina a guanina en la posición 263 y la inserción de una citosina en la posición 315. (Fig. 5).

La secuencia obtenida presenta una transición en la posición 16298 de timina a citosina, este haplotipo podría pertenecer al haplogrupo V [14] (aunque no es descartable que también pudiera ser asignada a un haplogrupo H). Entre los diez haplogrupos más comunes en individuos Caucásicos [15] (H, T, J, K, U, I, V, W, M, X) el haplogrupo V es uno de los que con menos frecuencia se presentan (1,6 %) [14]. Las secuencias obtenidas de las muestras indubitadas procedentes de familiares (saliva y pelo) coincidieron base a base con la obtenida de las muestras de fémur. Estos resultados pueden ser observados en la Tabla 2 y en la Fig. 5. La secuencia consensuada obtenida fue por último cotejada con las secuencias de ADNmt del personal del laboratorio no obteniéndose coincidencia alguna.
La secuencia final obtenida fue 16298C, 263G, 315.1C. Se realizó una consulta en una base de datos de 2192 individuos caucásicos, obteniéndose otra secuencia en dicha base de datos con idénticas discrepancias con respecto a la CRS. La frecuencia (p) de dicha secuencia fue de 0,0013 y para el cálculo del LR planteamos las siguientes hipótesis:
H0 = Probabilidad de obtener las secuencias del hueso y la muestra de referencia dado que el cadáver del que procede el hueso y el familiar que aporta muestra de referencia pertenezcan al mismo linaje materno.
H1 = Probabilidad de obtener las secuencias del hueso y la muestra de referencia dado que el cadáver del que procede el hueso y el familiar que aporta muestra de referencia no pertenezcan al mismo linaje materno.
LR = H0 / H1
El LR obtenido fue de 735. Este valor indica el número de veces que es más probable que obtengamos las secuencias descritas para el fémur y las muestras de referencia si dichas muestras son aportadas por personas emparentadas por vía materna frente a que se obtengan dichos resultados de secuencia si las personas que aportan dichas muestras no están emparentadas por vía materna.
Los resultados que aquí se presentan confirman una vez más, tal y como han publicado autores anteriores [17], que el análisis del ADN mitocondrial se convierte en una estrategia adecuada para llevar a cabo la identificación de cadáveres de cierta antigüedad o de aquellos casos en los que solo se dispone de parientes lejanos emparentados por vía materna. Esta última circunstancia es la que concurre en el caso que aquí se describe, puesto que no se disponía de más familiares que una sobrina.
Cuando se dispone de progenitores o descendientes directos (hijos/as) de la persona que se intenta identificar y además es posible obtener una cantidad y calidad adecuada de ADN nuclear para llevar a cabo un análisis de polimorfismo genético mediante marcadores nucleares (STRs), la opción del análisis de ADN nuclear es la elegida, ya que los índices de probabilidad aumentan extraordinariamente si se compara con los que se obtienen mediante análisis de ADNmt. Las bases de datos de ADNmt son bastante limitadas en cuanto al número de datos, y esta circunstancia explica los bajos índices de probabilidad que se obtienen cuando trabajamos con la opción del ADNmt. Actualmente se trabaja activamente desde el laboratorio de Medicina Legal de Innsbruck (Austria) en la creación de una gran base de datos mundial denominada EMPOP (EDNAP mtDNA population database) [18] que tiene por objetivo recopilar secuencias de ADNmt a nivel mundial y ofertar así a la comunidad forense una herramienta fundamental en el tratamiento estadístico de las muestras analizadas mediante ADNmt.
Si bien es cierto que los análisis de cuantificación de ADN nuclear de las muestras de fémur revelaron la presencia de ADN nuclear en muy escasa cantidad (inferior a 156 pg) es posible que dichas muestras pudieran, empleando las estrategias de amplificación adecuadas, generar perfiles genéticos interpretables. En la actualidad en nuestro laboratorio se están llevando a cabo distintos ensayos con el fin de poder obtener resultados para distintos short tandem repeats (STRs) nucleares. El estado de integridad del ADN de los restos óseos depende de diferentes variables ligadas a las condiciones medio ambientales de la zona donde se encuentre la fosa, de manera que es posible esperar, dependiendo de su procedencia, distinto estado de integridad del material genético. No obstante, pensamos que sería una buena aproximación poder determinar si con restos óseos de una antigüedad aproximada de 65 años es posible realizar análisis del ADN nuclear, puesto que en algunas ocasiones sí que se puede disponer de descendientes directos vivos o exhumados.
El cumplimiento de todos los requisitos impuestos en nuestro laboratorio para la aceptación de resultados finales garantiza la fiabilidad de los resultados y así mismo podemos afirmar que los grandes avances tecnológicos, centrados básicamente en los secuenciadores automáticos y software de análisis han facilitado enormemente la secuenciación del ADNmt, labor que hace tan solo unos pocos años resultaba terriblemente tediosa y no ausente de errores.
AGRADECIMIENTOS:
Los autores desean mostrar su agradecimiento a los médicos forenses Dr. Santiago Crespo Alonso, Dr. Gabriel Font Valserchi, Dra. Mercè Subirana Doménech, Dr. Josep Manel Tortosa López y Dr. Queral Solè Barjau por el laborioso y exhaustivo trabajo de campo realizado.
BIBLIOGRAFÍA:
- Bodenhagen D, Clayton DA. The number of mitochondrial deoxiribonucleic acid genomes in mouse L and human HeLa cells. J Biol Chem 1974; 249:1991-1995.
- Brown WM, George M and Wilson AC. Rapid evolution of animal mitochondrial DNA. Proc Natl Acad Sci USA 1979; 76: 1967-1971.
- Cann RL, Stoneckey M Wilson AC. Mitochondrial DNA and Human Evolution. Nature 1987; 325: 31-36
- Giles RE, Blanc H, Cann HM, Wallace DC. Maternal inherence of human mitochondrial DNA. Proc Natl Acad Sci USA 1980; 77: 6715-6719.
- Gill P, Ivanov PL, Kimpton C, Piercy R, Benson N, Tully G, et al. Identification of the remains of the Romanov family by DNA analysis. Nature Genetics. 1994; 6, 130-135.
- Jehaes E, Pfeiffer H, Toprak, Decorte R, Brinkmann B, Cassiman J-J. Mitocondrial DNA analysis of the putative heart of Louis XVII son of Louis XVI and Marie-Antoinette. Eur J Hum Genet 2001; 9: 185-190.
- Anderson S, Bankier AT, Barrell BG, Bruijn MHK de, Coulson AR, Douin IC et al. Sequence and organization of the mitochondrial genome. Nature 1981; 290: 457-465.
- Wilson MR, DiZinno JA, Polanskey D, Reploge J, Budowle B. Validation of mitochondrial DNA sequencing for forensic casework analysis. Int J Legal Med 1995; 108: 68-74.
- Carracedo A, Bär W, Lincoln P, Mayr W, Morling N, Olaisen B, et al. DNA Commission of the International Society for Forensic Genetics: guidelines for mitochondrial DNA typing. Forensic Sci Int 2000; 110: 79-85.
- Tully G, Bär W, Brinkmann B, Carracedo A, Gill P, Parson W et al. Considerations by the European DNA profiling (EDNAP) group on the working practices, nomenclature and interpretation of mitochondrial DNA profiles. Forensic Sci Int 2001; 124: 83-91.
- Holland MM, Parsons TJ. Mitochondrial DNA sequence analysis validation and use for forensic casework, Forensic Sci Rev 1999; 11: 21-50.
- Balding DJ, Nichols RA. DNA profile match probability calculation: how to allow for population stratification, relatedness, database selection and single bands. Forensic Sci Int 1994; 64: 125-140.
- Evett IW, Weir BS. Interpreting DNA evidences- Statistical Genetics for Forensic Scientists, Sinauer, MA. 1998 142-146.
- Helgason A, Hickey E, Goodacre S, Bosnes V, Stefansson K, Ward R, et al. MtDNA and the Islands of the North Atlantic: estimating the proportion of Noorse and Gaelic ancestry. Am J Hum Genet 2001; 68: 723-737.
- Torroni A, Huoponen K, Francalacci P, Petrozzi M, Morelli L, Scozzari R et al. Classification of European mtDNAs from an analysis of three European population. Genetics 1996; 114: 1835-1850.
- Allard WA, Miller K, Wilson M, Monson K, Budowle B. Characterization of the Caucasian Hoplogroups present in the SWGDAM forensic mtDNA dataset for 1771 human control region sequences. J Forensic Sci 2002; 47: 1215-1223.
- Holland MM, Fisher DL, Mitchell LG, Rodriguez WC, Canik JJ, Merril CR, et al. Mitochondrial DNA sequence analysis of human skeletal remains from the Vietnam War. J Forensic Sci 1993; 38: 542-553.
- Parson W, Brandstätter A, Alonso A, Brandt N, Brinkmann B, Carracedo A et al. The EDNAP mitochondrial DNA population database (EMPOP) collaborative exercises: organisation, results and perspectives. Forensic Sci Int 2004; 139: 215-226.
Correspondencia:
Dr. Manuel Crespillo Márquez.
Servicio de Biología. Instituto Nacional de Toxicología y Ciencias Forenses.
Calle Merced, nº 1. 08002 Barcelona. Telf. 93 317 40 61. Fax 93 318 25 30.
E-mail: manuel.crespillo@mju.es