Atlas de Patología Forense
Muerte súbita en un joven con Síndrome de Marfan
Sudden death in a young man with Marfan's Syndrome.

Cuad Med Forense. 2005; 11(42):317-325
RESUMEN
El síndrome de Marfan es un trastorno del tejido conectivo con grados de expresión clínica variable. Las manifestaciones clínicas más importantes de este síndrome suelen localizarse a nivel ocular, esquelético y cardiovascular, siendo estas últimas las que con mayor frecuencia conducen a la muerte. Las complicaciones cardiovasculares se presentan principalmente a nivel valvular y aórtico; en concreto a este último nivel es común observar áreas de degeneración quística de la capa media de la aorta (medionecrosis quística), caracterizada por la presencia de apoptosis y por la pérdida de células musculares lisas. Aunque el sustrato biológico específico es desconocido, se ha atribuido a la existencia de un aumento en la síntesis de ácido hialurónico, un aumento en la solubilidad del colágeno y de la elastina, alteraciones en la cantidad relativa de colágeno tipo I y III y más recientemente la presencia de cadenas de colágeno-alfa2.
El interés de este cuadro en Patología forense radica en su posible presentación en forma de muerte súbita sin ningún tipo de clínica previa. Resulta por tanto fundamental identificar cuidadosamente todos los hallazgos necrópsicos para poder relacionarlos con este síndrome. La cuestión posee además un interés epidemiológico al objeto de comunicar a la familia del fallecido aquella información que sea de importancia médica para otros miembros de la misma.
Exponemos un caso de estas características en un varón joven, sin patología previa conocida que falleció súbitamente a la salida de una discoteca. El estudio necrópsico reveló una disección aórtica en su porción extrapericárdica como causa responsable de la muerte. El caso se ilustra además con el correspondiente estudio histopatológico específico.
Palabras clave: Síndrome de Marfan, Muerte súbita, Disección aórtica, Patología forense.
ABSTRACT
Marfan´s syndrome is a disorder of the connective tissue with a variable clinical expression. Clinically the most important manifestations are the ocular, skeletal and cardiovascular ones, which frequently lead to death. Cardiovascular complications involve mainly the valves and the aorta, in fact at this level is common finding cystic medial degeneration areas, characterized by apoptosis and by the loss of vascular smooth muscle cells. Although the specific biological defect is unknown, it has been thought to be an increase of the hialuronic acid synthesis, an increase of the collagen and elastine solubility, the alteration in the ratio collagen type-1 and type-3 and more recently, the presence of collagen-alpha2 chains.
The interest of this syndrome in forensic pathology is that it can be present as a sudden death without any previous manifestation, being very important to identify carefully all the autopsy findings to relate them with this syndrome. It has also an epidemiologic interest with the aim of communicating the information that may be of medical importance to other family´s members.
We report a case with these characteristics in a young man without previous symptoms who died suddenly at the disco's exit. The macroscopic examination showed aortic dissection in the extrapericardical portion, considered as cause of the death. The case is also illustrated with the specific histopathologic study.
Key words: Marfan's syndrome, Sudden death, Aortic dissection, Forensic pathology.
INTRODUCCIÓN:
El síndrome de Marfan es un trastorno hereditario del tejido conectivo que debe su nombre a un pediatra parisino (Marfan) quien publicó la primera descripción en 1896.
El cuadro se hereda de forma autosómica dominante, con una penetrancia variable y sin preferencias étnicas ni raciales.
Su prevalencia es de 4-6 casos/100.000 habitantes [1] y en la expresividad de la enfermedad influye de manera importante la edad de los progenitores, especialmente en aquellos casos de mutaciones espontáneas. Para ciertos autores, aproximadamente un 15% de los casos no presentan una historia familiar previa [2].
En términos generales, esta patología se puede clasificar en forma clásica y formas parciales, para referirnos a aquellos casos en los que algunas de las alteraciones orgánicas están disminuidas o ausentes. La mayoría de los autores consideran necesaria la presencia de más de dos signos clásicos para el diagnóstico, mientras que para otros sería suficiente la presencia de alteraciones a nivel ocular y una historia familiar positiva [3].
Las manifestaciones clínicas típicas se localizan a nivel ocular, con ectopia lentis, miopía o desprendimiento de retina; a nivel esquelético, con deformidades torácicas, aracnodactilia, escoliosis o hiperlaxitud articular; pero sin duda son las manifestaciones cardiovasculares las que acarrean las mayores complicaciones. Estas complicaciones abarcan la insuficiencia aórtica y mitral, aneurismas y disección aórtica, así como endocarditis bacteriana [2, 4, 5]. También se han descrito, aunque con menor frecuencia, neumotórax espontáneos, anomalías pulmonares congénitas y hernias inguinales con tendencia a la recidiva tras la reparación quirúrgica [1].
El diagnóstico debe establecerse en la infancia o en la juventud y el seguimiento ha de ser multidisciplinar, comprendiendo revisiones oftalmológicas anuales, revisiones semestrales por el traumatólogo, en aquellos casos de escoliosis y en adultos con deformidades progresivas, y control por el cardiólogo, mediante ecocardiogramas anuales, y por el cirujano vascular. Además es frecuente que estos pacientes acudan a ginecólogos y genetistas, dado el carácter hereditario de este trastorno y del mayor riesgo para la gestante con este síndrome.
Por lo que respecta al tratamiento de las complicaciones cardiovasculares, en pacientes con cambios aórticos incipientes se recomienda el uso de propanolol y de reserpina dada la acción inotrópica negativa de estos agentes, reduciendo la contractilidad miocárdica y la presión pulsátil que ejerce el flujo sanguíneo sobre la aorta dilatada.
En fases más avanzadas el tratamiento de elección suele ser quirúrgico. En pacientes con patología valvular aórtica y aneurisma de aorta torácica ascendente la tendencia es el remplazamiento total de la válvula y de la porción tubular aórtica, ya que tanto la raíz aórtica como la válvula y el anillo aórtico suelen estar difusamente afectados. La sustitución completa es necesaria no solo para restaurar la situación sino también para prevenir recurrencias.
En el estudio de la historia natural del síndrome de Marfan, Murdoch y cols establecieron en 32 años la esperanza de vida media de estos pacientes (2), siendo la causa de muerte en el 95% de los casos las complicaciones cardiovasculares, presentándose en forma de muerte súbita con gran frecuencia [5].
En estos casos, las principales causas de los fallecimientos son las complicaciones de la pared aneurismática de la aorta ascendente, la insuficiencia de las válvulas mitral y/o aórtica, la endocarditis bacteriana subaguda, la disección aórtica y la ruptura de un aneurisma aórtico [2,5].
El hallazgo más frecuente en las dilataciones aórticas es la degeneración de la media, denominada clásicamente «medionecrosis quística». Este término se usaba para describir una situación en la que se combinaba una necrosis de las células musculares lisas y una degeneración mucoide de la media, sin embargo este término ha sido criticado por su inespecificidad y por la ausencia de quiste, siendo utilizado actualmente para hacer referencia a la mera presencia de basofilia en la media. Se ha propuesto incluso una explicación al proceso, según la cual sería el común resultante de la acción de las fuerzas hemodinámicas y del continuo proceso de daño-reparación que ocurre en el envejecimiento de la aorta [6].
Estos mismos procesos ocurrirían en el síndrome de Marfan, pero dada la alteración del tejido conectivo subyacente que existe en esta patología, el daño es más severo cuanto más temprana sea la edad del paciente [4,5].
A pesar de que el defecto en el sustrato biológico específico es desconocido, se han descrito varias anomalías bioquímicas tanto en la piel como en el tejido aórtico de pacientes con síndrome de Marfan que incluyen aumento en la síntesis de ácido hialurónico, aumento de la solubilidad del colágeno y de la elastina, alteración en las cantidades relativas de colágeno tipo 1 y tipo 3, anomalías en los enlaces y, más recientemente, se ha descrito una cadena de colágeno-alfa2 en un paciente con síndrome de Marfan [3,7,8].
Una de las características de la degradación quística de la media es el alto índice de apoptosis y la pérdida de células musculares lisas vasculares que podría estar en relación con la activación del receptor-gamma de la proliferación del peroxisoma, un factor de trascripción de la superfamilia de los receptores nucleares [9].
Por otra parte, algunos investigadores han descrito en los aneurismas de aorta torácica una hiperplasia de las células musculares lisas vasculares, con conservación de la densidad celular y aumento de la masa de la capa media; así como un incremento en la degradación de la matriz. Este último hallazgo también se encuentra en aneurismas de la aorta abdominal, sin embargo el primero puede ser una respuesta adaptativa inicial para minimizar el aumento de presión sobre la pared resultante de la dilatación vascular [10].
En el presente artículo se expone un caso de muerte súbita asociado a este síndrome. La cuestión posee un elevado interés cuando su presentación es en forma de muerte súbita sin ningún tipo de clínica previa en una persona joven, como es el caso. Dado el carácter hereditario de esta alteración, en los casos de sujetos no diagnosticados previamente, corresponde al médico forense la tarea de identificar cuidadosamente todos los hallazgos necrópsicos y comunicar a la familia del fallecido, aquellos que sean de importancia médica para otros miembros de la misma.
DESCRIPCIÓN DEL CASO:
El cadáver correspondía a un varón de raza caucasoide, de 21 años de edad, soltero, jugador de baloncesto, 186 cm de longitud y 78 kilogramos de peso con un índice de masa corporal de 22.55. El joven falleció súbitamente a la salida de una discoteca. En los momentos previos a la muerte presentó dolor torácico y molestias inespecíficas (sensación de mareo y malestar general), según informaron quienes le acompañaban.
En sus antecedentes no constaban datos de enfermedades importantes, intervenciones quirúrgicas previas ni tratamientos farmacológicos.
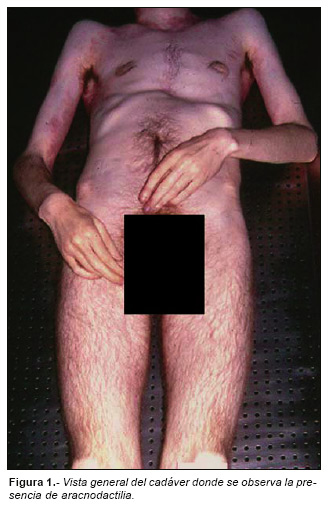
Externamente presentaba elevada estatura, extremidades extremadamente largas especialmente en los antebrazos y en los muslos (dolicochosenamelia), manos delgadas y dedos alargados (aracnodactilia) (Fig. 1), dolicocefalia y depresión infundibuliforme de la cara anterior del tórax (pectus excavatum) (Fig. 2). En la familia no existían otros miembros que tuvieran este mismo aspecto. Vulgarmente era considerado «un larguirucho».

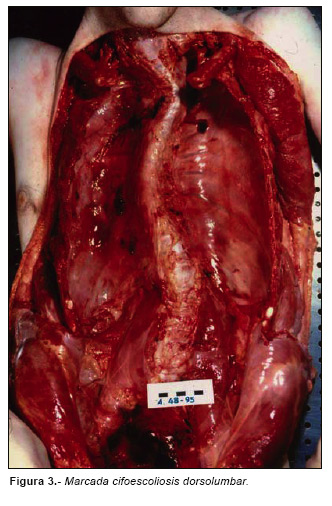
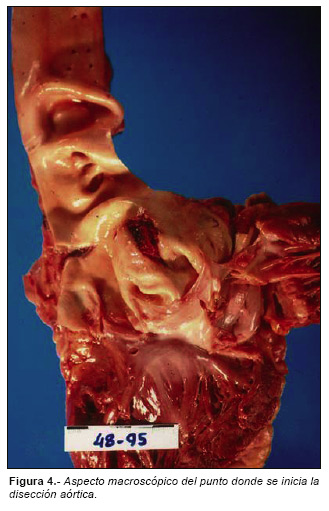
El examen interno mostraba deformidad de la columna vertebral en forma de pronunciada cifoescoliosis (Fig. 3). Se encontró además un desgarro de 2 cm. en la porción ascendente intrapericárdica de la aorta torácica (Fig. 4) con disección de sus capas y formación de un voluminoso hematoma rodeando la raíz de los grandes vasos (Fig. 5), así como un voluminoso hemopericardio (375 cc). El corazón pesaba 386 gr con un ventrículo izquierdo de 16 mm de espesor.

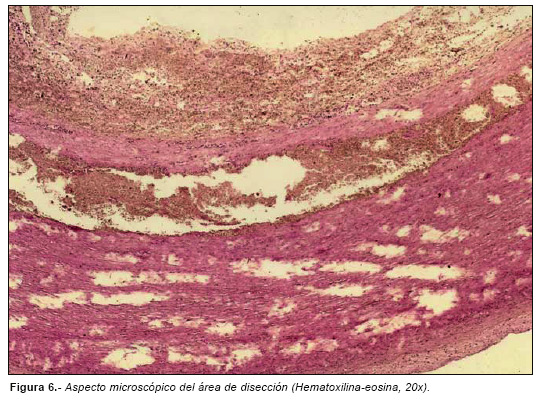
El estudio microscópico ofreció los siguientes datos de interés: disección de la capa media aórtica (Fig. 6) con áreas de degeneración quística (Fig. 7) con depleción y fragmentación de fibras elásticas (Fig. 8), asociada a un incremento de las fibras de colágeno y de la vascularidad de la adventicia.


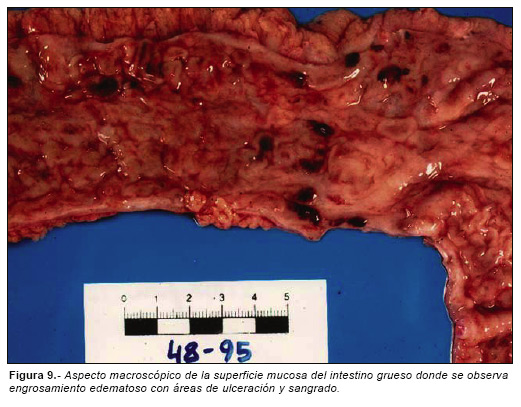
Además de los hallazgos descritos, el cadáver presentaba un marcado engrosamiento edematoso e hiperhémico de la mucosa del intestino grueso, adoptando un aspecto «empedrado» salpicado por zonas hemorrágicas (Fig. 9). Se observó además infiltrado linfoplasmocitario transmural con abundantes granulomas de células gigantes, sin necrosis central. Se apreciaron además zonas de erosión mucosa superficial y formación de criptas en cuya base se observaban acúmulos linfoides, hallazgos compatibles con enfermedad de Crohn. El estudio toxicológico fue negativo.
DISCUSIÓN:
En el síndrome de Marfan suele existir una historia familiar previa de la enfermedad, sin embargo nuestro caso se engloba en el pequeño porcentaje del 15% descrito por autores como Bain [3] en los que se presenta como resultado de una mutación espontánea.
Al contrario de lo que es habitual, no se detectaron alteraciones oculares, valvas flácidas (flappy valves) ni cardiomegalia, ni defectos del tabique interauricular, interventricular ni coartación aórtica.
Por su parte, la ausencia de luxación del cristalino (ectopia lentis) es considerada normal por autores como Robins [11], sin embargo otros como Isselbacher [12] elevan su prevalencia hasta el 75% de los casos.
Desde el punto de vista epidemiológico, nuestro caso no se ajustaba a lo descrito por Pyeritz [1] según el cual la muerte sobreviene hacia la mitad de la vida (expectativa de vida normal), ni tampoco cumplía el criterio descrito por Murdoch [2] quien opina que el fallecimiento ocurre mayoritariamente entre los 30 y los 40 años de edad.
Sin embargo se ven refrendadas las opiniones de Robins [11] y aún más la de Patton [13] al afirmar que las complicaciones cardiovasculares representan los rasgos más amenazadores de este trastorno con un rango de mortalidad entre el 30% [11] y el 90% [13], además del hecho de que estas frecuentemente ocurren súbitamente sin alteraciones clínicas previas [4].
De igual forma, se confirma la aseveración de Maron (14) toda vez que la causa de la muerte en nuestro caso fue la disección aórtica. El citado autor considera que un 7% aproximadamente de la muerte súbita ocurrida en deportistas menores de 30 años se debe a esta causa en particular.
Bain [3] atribuye este fenómeno a que el ejercicio físico probablemente provoque un aumento en la presión sanguínea lo suficientemente importante como para romper la capa media en una aorta ya debilitada y dilatada.
A nivel aórtico encontramos un desgarro en la porción ascendente intrapericárdica, concordando estos hallazgos con los de Roberts [5], en cuyos estudios la frecuencia de presentación de desgarros en la porción aneurismática de la aorta es del 50% y aún más con los de Dickens [15] quien eleva esta frecuencia al 95%. El mismo autor evidenció que la aorta de estos pacientes podía presentar hemorragia intramural incluso cuando el mecanismo de disección no está claro. En nuestro caso la presencia de infiltrado leucocitario rodeando el desgarro sugiere que el mismo, aunque agudo, fue precedido durante algún tiempo de un progresivo sangrado.
Tal y como afirma Cigarroa [16] la disección tuvo lugar en la porción ascendente de la aorta asentando sobre la porción dilatada, hecho observado igualmente por Gillan [8].
Otras dilataciones aneurismáticas han sido descritas [15] en aorta torácica descendente y abdominal, arterias pulmonares, vasos cerebrales, ductus arterioso y arterias coronarias; descartándose en nuestro caso hallazgos a estos niveles.
En el estudio microscópico los signos encontrados concuerdan con los de Dickens [15] al evidenciarse a nivel de la pared aneurismática una fragmentación severa, atrofia y pérdida de fibras elásticas, aumento del colágeno así como de material mucopolisacárido.
Este autor también describió un aumento en la vascularización de la media, sin embargo nosotros hallamos este fenómeno solo a nivel de la adventicia.
Dickens [15] considera que la causa de la disección es la marcada necrosis quística de la media con fragmentación elástica, sin embargo el papel de esta lesión no está claro y únicamente es significativo en el síndrome de Marfan, donde aparece en el 82% de los fallecimientos con aneurisma disecante, coincidiendo con los hallazgos de nuestro caso; frente al 19% de los casos no-Marfan. Este podría ser un factor significativo, unido al hecho de la ausencia de historia previa de hipertensión arterial.
Tampoco había evidencias de otras condiciones que han sido ocasionalmente asociadas a la disección como la estenosis de la válvula aórtica o la válvula aórtica bicuspídea [15].
Otra cuestión que pudiera crear controversias sería la existencia de homocistinuria, algunos pacientes presentan un aumento de hidroxiprolina en orina, sin embargo según informaciones familiares se descartó su posible presencia durante la niñez. En las sucesivas revisiones pediátricas nunca se había observado esta anomalía.
Al final del estudio macro y microscópico pudimos concluir que se trataba de un verdadero síndrome de Marfan, hecho que fue puesto en conocimiento de la familia para posible estudio de sus miembros.
Finalmente merece un comentario la presencia de datos de enfermedad de Crohn. En la literatura médica revisada no hemos encontrado ninguna referencia a esta asociación como un síndrome de características propias, por lo que consideramos que se trataba de una asociación casual.
BIBLIOGRAFÍA:
- Pyeritz RE, McKusick VA: The Marfan syndrome: diagnosis and management. N Engl J Med 1979; 300: 772.
- Murdoch JL, Walker BA, Halpern BL, Kuzuma JW, McKusick VA: Life expectancy and causes of death in the Marfan syndrome. N Engl J Med 1972; 286: 804.
- Bain MA, Zumwalt RE, Van der Bel-Kahn J: Marfan syndrome presenting as aortic rupture in a young athlete: sudden unexpected death? Am J For Med Path 1987; 8: 334.
- Crawford ES: Marfan’s syndrome: broad spectral surgical treatment cardiovascular manifestations. Ann Surg 1983; 198: 487.
- Roberts WC, Honing HS: The spectrum of cardiovascular disease in the Marfan’s syndrome: a clinico-morfologic study of 18 necropsy patients and comparison to 151 previously reported necropsy patients. Am Heart J 1982; 104: 115.
- Schlatmann TJM, Becker AE: Pathogenesis of dissecting aneurysm of aorta. Comparative histopathologic study of significance of medial changes. Am J Cardiol 1977; 39:21.
- Buchanan R, Wyatt GP: Marfan’s syndrome presenting as an intrapartum death. Arch Dis Child 1895; 60:1074.
- Gillan JE, Costigan DC, Keeley FW, Rose T, Cutz E, Rose V: Spontaneus dissecting aneurysm of the ductus arteriosus in an infant with Marfan’s syndrome. J Pediatrics 1984; 105: 952.
- Sakomura Y, Nagashima H, Aoka Y, Sukuta A, Kurosawa H, Nishikawa T, Kasanuki H: Expression of peroxisome proliferator-activated receptor-gammain vascular smooth muscle cells is upregulated in cystic medial degeneration of annuloaortic ectasia in Marfan syndrome. Am Heart Association 2002; 106:I-259.
- Tang PCY, Coady MA, Lovoulos C, Dardik A, Aslan M, Elefteriados JA, Tellides G: Hyperplasic cellular remodelling of the media in ascending thoracic aortic aneurysm. Am Heart Association 2005; 112: 1098.
- Robbins Sl, Cotran RS, Kumar V. Patología estructural y funcional. Interamenricana (ed). Saunders Company, Madrid, 1990. Vol I, p. 143.
- Isselbacher KJ, Martin JB, Braundwald E et al. Harrison’s Principles of Internal Medicine, 30th edition, Mcgraw-Hill (ed), New York, 1994; p. 2116.
- Patton DJ, Galliani CA, Johnson WH Jr, Hedlung GL. Sudden death in Marfan syndrome. Am J Roentgenol 1995; 165: 160.
- Maron BJ, Epstein SE, Roberts WC. Causes of sudden death in competitive athletes. J Am Col Cardiol 1986; 7: 204.
- Dickens P, Khoo US. Sudden death associated with bloodless aortic dissection. Forensic Sci Int 1993; 149:155.
- Cigarroa JE, Isselbacher EM, Desanctis RW, Eagle KA. Diagnostic imaging in the evaluation of suspected aortic dissection. N Engl J Med 1993; 328: 35.
Correspondencia:
Dr. José Blanco Pampín.
Instituto de Medicina Legal de Galicia. Servicio de Patología Forense.
c/ Viena s/n, 4ª planta. 15705. Santiago de Compostela (A Coruña).
E-mail: cmpampin@usc.es